(or Phenylketonuria type 2)
See also Phenylketonuria
1. Forms with hyperphenylalaninemia or type 2 phenylketonuria
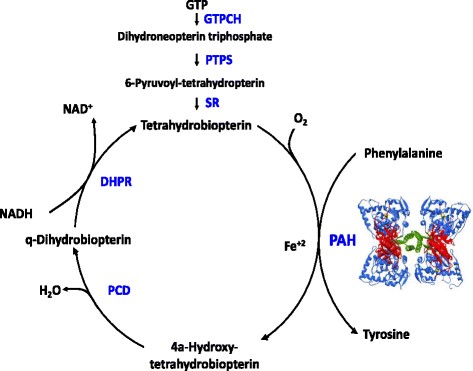
Autosomal recessive transmission of a mutation of one of the genes ensuring the synthesis or regeneration of tetrahydrobiopterin (BH4), a cofactor of the phenylalanine hydroxylase, of the tyrosine hydroxylase, of the tryptophan hydroxylase (2 isoforms), of the alkylglycerol mono-oxygenase and of the NO synthetase (3 isoforms): they are GCH1 (GTP cyclohydroxylase I on 14q22.2) [MIM 233 910], PTS (6-pyruvoyl-tetrahydropterine synthase on 11q23.1)(54 %) [MIM 261 640], PCBD (pterine-4α-carbinolamine dehydratase on 10q22.2 [MIM 264 070], or QDPR (dihydropteridine reductase on 4p15.32)(33 %)[MIM 261 630].
The deficiency in BH4 due to a mutation of one of those genes causes a form of phenylketonuria (1-2 % of cases) as, in addition to hyperphenylalaninemia, the failure of all those enzymes results in a deficiency in the monoaminergic neurotransmitters: dopamine, serotonin and NO.
In developed countries, phenylketonuria is part of systematic neonatal screening a few days after birth (Guthrie's test). In case of a pathological Phe blood level, a response test to oral BH4 intake is performed: 20 mg/kg/ d for 48 hours. If the Phe blood level decreases for more than 30 % of the initial level, a treatment with BH4 and a diet (often less severe than for the classic type 1 phenylketonuria) is proposed.
With no special diet, this form of hyperphenylalaninemia leads to:
- hypotonia and severe mental retardation
- dystonia, uncoordinated movements, tremor at rest, extrapyramidal rigidity with bradykinesia
- behavioral disorders: hyperactivity, attention disorders, anxiety, compulsive obsessionaldisorders, psychosis
- sleep disorders, excessive salivation
- occasional flexion spasms and epilepsy
- episodes of hyperthermia
A hypomelanosis (pale skin, blond hair, blue eyes, eczema) can also be present, as well as a characteristic so-called 'musty odor'.
In case of pterin-4α-carbinolamin dehydrase deficiency, hypomagnesemia and early diabetes (MODY3 type) can be present. In case of hyperphenylalaninemia without mutation of the phenylanine hydroxylase nor anomaly of the tetrahydropterin metabolism, it is necessary to search for a mutation of the DNAJC12, a chaperone protein contributing to the deployment of the phenylalanine hydroxylase.
Three possible clinical presentations:
- asymptomatic: hyperphenylalaninemia discovered thanks to Guthries test, allowing early diagnosis and timely treatment
- symptomatic (neurologic deterioration) despite an appropriate diet: wrongly diagnosed as isolated hyperphenylalaninemia (type 1)
- symptomatic without any diet: falsely negative test at birth ?
In addition to a low phenylalanine diet associated with substitutes of amino acids, the disease responds favorably to:
- tetrahydrobiopterine (BH4), the cofactor of phenylalanine hydroxylase, allowing a less strict diet. The dose (sapropterin dihydro or Kuvan®) varies from 5 to 20 mg/kg/day in 2-3 doses. However, supplements of BH4 have to be avoided in case of dihydropteridin reductase deficiency as there is a risk of excess of dihydrobiopterin..
- folic acid: 15 mg/day
- 5-hydroxytryptophan: from 1 to 10 mg/kg/day in 4 doses except in case of pterin-4á-carbinolamin dehydratase or guanosine triphosphate cyclohydrolase deficiency
- L-Dopa associated with a dopa-decarboxylase inhibitor: 1-2 mg/kg/day in 4 doses
and, in some cases, to
- entacapone (catechol-o-methyl-transferase inhibitor): 15 µg/kg/day in 2-3 doses
- selegiline (MAO-B inhibitor): from 0.1 to 0.25 mg/kg/day in 3-4 doses (not to be co-administered with 5-hydroxytryptophan : risk of serotoninergic syndrome)
- pramipexole (a dopamine receptor agonist): 6 to 35 µg/kg/day in 2 doses.
Warning: Aspartame and gelatin are to be avoided in children with hyperphenylalaninemia. Aspartame is the methyl ester of phenylalanine aspartate that splits into 2 amino acids, aspartic acid and phenylalanine, and methanol. Gelatin, which often enters in the composition of oral capsules, is particularly rich in proteins (86 g / 100 g of product). Aspartame and gelatin are frequent components of oral drugs. Therefore, great care should be taken to avoid medicines containing these substances. The amount of phenylalanine brought by both substances, however, remains low. The drug can be administered carefully with monitoring of the blood level and decreasing concomitantly the amount of food-borne phenylalanine (involvement of a dietician is necessary) if a treatment is really necessary and that there is no alternative without aspartame or without gelatin. It is always useful to check the composition of a drug (list of excipients) in a formulary.
2. Forms without hyperphenylalaninemia
Two forms:
- autosomal dominant transmission of a mutation of the GCH1 gene (14q22.2) causing a deficiency in guanosine triphosphate cyclohydrolaseI [MIM 128 230]: it is the most frequent cause of dopa responsive dystonia, a syndrome characterized by fluctuating dystonia (getting worse along the day), also called autosomal dominant Segawa syndrome (DYT5a) (see this term)
- automal recessive transmission of a mutation of the PR gen (2p13.2), leading to a deficiency in sepiapterin reductase [MIM 612 716]
Anesthetic implications:
In the forms of hyperphenylalaninemia, the necessary precautions to avoid increasing Phe blood levels should be applied, such as:
- advice from the endocrinologist and knowing the patient's usual of Phe blood level
- in adults, check cardiac and renal function
- any infection increases the Phe blood level and requires an adaptation of the Phe intake
- avoid any protein catabolism: shorten as much as possible the preoperative fasting time and ensure adequate glucose intake perioperatively; check blood glucose regularly.
- due to the risk of vitamin B12 deficiency if the diet is not adequately supplemented, it is wise to avoid N2O.
- in case of parenteral feeding, use a Phe-free IV solution.
- colloids based on gelatins: although their elimination is mainly in an unchanged form in the urine, they should be used with caution.
- in case of risk of blood ingestion (oropharyngeal surgery: tonsillectomy, adenoids, cleft palate, stomatology) it is useful to place a pharyngeal packing and empty the stomach before the end of the procedure as swallowed blood is rich in protein.
In all forms, consideration should also be given to drug interactions between the anesthetic agents and the patient's chronic treatment:
- L-dopa: chronic intake of L-dopa may result in orthostatic hypotension and decrease the response to the indirect vasopressors such as ephedrine; it is therefore better to use titrated doses of direct vasopressors. Central dopamine antagonists such as metoclopramide and butyrophenones should also be avoided.
- pramipexole (dopamine receptor agonist)
- 5-hydroxytryptophan
- entacapone (a COMT inhibitor) that increases the sensitivity of the response to the direct vasopressors
- selegiline (type B MAO inhibitor) in 3-4 doses: in principle no interaction with the anesthetic agents for doses less than 10 mg/d but it could act like an type A IMAO at higher doses.
It is not known whether setrons (5HT3 inhibitors) can be used as antiemetics. It is preferable to avoid tramadol because of its monoaminergic mode of action. Avoid trimethoprim / sulfametroxazole.
In addition, there is a risk of postoperative of behavioral disorders and hyperthermia.
References:
- Dal D, Celiker V.
Anaesthetic management of a strabismus patient with phenylketonuria.
Paediatr Anaesth 2003; 13: 740-1. et correspondance: Pediatr Anesth 2004; 14: 701-2.
- Feillet F, Bonnemains C.
La phénylcétonurie : nouveaux traitements.
Arch Pédiatr 2013 ; 20 : 1165-8.
- Wyatt SS, Gill RS.
An absolute contraindication to nitrous oxide.
Anaesthesia 1999 ; 54 : 307.
- Walter JH, Lachmann RH, Burgard P.
Hyperphenylalaninaemia,
In Inborn Metabolic Diseases, 5th edition, Saudubray, van den Berghe, Walter, Springer 2011, p 251-64.
- Opladen T, Lopez-Laso E, Cortes-Saladelafont E, Pearson TS, Sivri HS et al.
Consensus guidelines for the diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies.
Orphanet J Rare Diseases 2020; 15:126
Updated: July 2020