[MIM 261 600]
(phenylpyruvic oligophrenia, phenylketonuria type I, Folling disease)
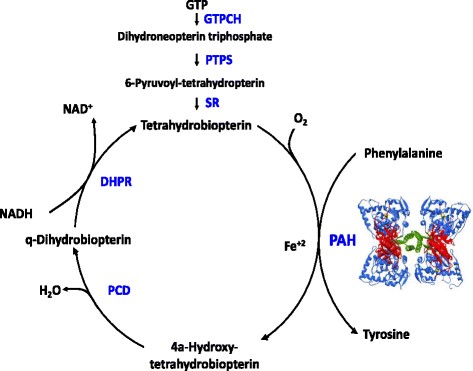
Prevalence: 1/18,000 but varies from 1/2.600 in Turkey to 1/50.000 in U.K. and 1/100.000 in Japan. Autosomal recessive inheritance: 98 % of cases are due to a mutation (more than 950 described mutations) of the PAH gene (12q23.2) coding for the phenylalanine hydroxylase involved in the synthesis of tyrosine, L-dopa and noradrenaline. A minority of cases (phenylketonuria type 2) is due to a mutation in one of the genes ensuring the synthesis or regeneration of tetrahydrobiopterin (BH4): GTPCH, PTPS, PCD, DHPR. (see tetrahydrobiopterin deficiency)
In developed countries, phenylketonuria is part of systematic screening a few days after birth (Guthrie's test). The lifelong treatment, which consists of a phenylalanine-poor diet associated with amino acids supplements supplements, is introduced if the blood levels of phenylalanine exceed 360 µmol/l. Target level: 120 to 360 µmol/L. Target level: 120 to 360 µmol/L before 12 years of age of during pregnancy, 120 to 600 µmol/L after 12 years of age.
The other causes of hyperphenylalaninemia have be excluded such as prematurity, liver failure, anomaly of the BH4 metabolism. A response test to the oral administration of BH4 is performed: 20 mg/kg/d for 48 hours. If the Phe blood level decreases for more than 30 % of the initial level, a treatment with BH4 and a less severe diet are proposed (see tetrahydrobiopterin deficiency). In case of hyperphenylalaninemia without mutation of the phenylanine hydroxylase nor anomaly of the tetrahydropterin metabolism, it is necessary to search for a mutation of the DNAJC12, a chaperone protein contributing to the deployment of the phenylalanine hydroxylase.
Without diet, the disease causes severe mental retardation (behavior disorders, psychoses, flexion spasms, epilepsy) hypomelanosis (fair skin, blond hair, blue eyes) and a characteristic so-called 'musty odor'.
These signs should be kept in mind when examining children born in countries where neonatal screening is not routinely performed. Great heterogeneity of the phenotype depends on the residual enzyme activity and the response to BH4 intake.
Four forms of hyperphenylalaninemia are distinguished according to Phe tolerance, i.e. the maximal daily dose of alimentary Phe keeps the Phe blood levels between 120 and 360 µmol/L:
- classical or severe: 250-350 mg daily Phe intake
- moderate: 350-400 daily Phe intake
- benign: 400-600 mg daily Phe intake
A new treatment makes it possible to treat these patients without any special diet: pegvaliase (Palynziq® BioMarin, Novato, CA), an injectable form of phenylalanine ammonia lyase, an enzyme that metabolizes blood phenylalanine and replaces the deficient activity of phenylalanine hydroxylase. Subcutaneous dose: 2.5 to 40 mg/week. Therapeutic goal: blood Phe level between 30 and 360 µmol/L.
Non-phenylketonuric hyperphenylalaninemia is present when the blood Phe level remains under 600 µmol/L with a normal diet. Those patients have normal development and behavior and the necessity to impose a diet is a controversial topic.
Warning: aspartame and gelatin are to be avoided in children with phenylketonuria. Aspartame is the methyl ester of phenylalanine aspartate that splits, in the body, in two amino acids, aspartic acid and phenylalanine, and methanol. Gelatin often enters in the composition of capsules and is particularly rich in proteins (86 g / 100 g of product). These two substances are often components of oral drugs. Therefore, care should be taken to avoid drugs containing these substances as adjuvants. However the quantity of phenylalanine brought by these substances remains low and if a treatment is really necessary and if there is no alternative without aspartame or gelatin, the drug can be administered but the phenylalanine blood level should be monitored and the amount of alimentary phenylalanine should be decreased (advice from a dietician).
It is always useful to verify the formulation of a drug (list of excipients) in a formulary.
PKU adults with a Phe-poor diet since birth can present with:
- an atherogenic dyslipidemia, risk of overweight and an increased rigidity of the arterial walls
- proteinuria and decreased glomerular filtration rate
Anesthetic implications:
- advice from the endocrinologist and knowing the patient's usual Phe blood level; the implications of a treatment by pegvaliase are not known yet.
- in adults, check cardiac and renal function
- any infection increases the Phe blood level and requires an adaptation of the Phe intake
- avoid any protein catabolism: shorten as much as possible the preoperative fasting time and ensure adequate glucose intake perioperatively; check blood glucose regularly.
- due to the risk of vitamin B12 deficiency if the diet is not adequately supplemented, it is wise to avoid N2O.
- in case of parenteral feeding, use a Phe-free IV solution.
- colloids based on gelatins: although their elimination is mainly in an unchanged form in the urine, they should be used with caution.
- in case of risk of blood ingestion (oropharyngeal surgery: tonsillectomy, adenoids, cleft palate, stomatology) it is useful to place a pharyngeal packing and empty the stomach before the end of the procedure as swallowed blood is rich in protein.
References :
- Dal D, Celiker V.
Anaesthetic management of a strabismus patient with phenylketonuria.
Paediatr Anaesth 2003; 13: 740-1. et correspondance: Pediatr Anesth 2004; 14: 701-2.
- Feillet F, Bonnemains C.
La phénylcétonurie : nouveaux traitements.
Arch Pédiatr 2013 ; 20 : 1165-8.
- Van Wegberg AMJ, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, Bosch AM, Burlina A et al.
The complete European guidelines on phenylketonuria: diagnosis and treatment.
Orphanet Journal of Rare Diseases 2017; 12:62
- Mahan KC, Gandhi MA, Anand S.
Pegvaliase: a novel treatment option for adults with phenylketonuria.
Curr Med Res Opin 2019;35:647-51. doi: 10.1080/03007995.2018.1528215.
- Cunningham A, Rohr F, Splett P, Mofidi S, Bausell H, Stembridge A, Kenneson A, Singh RH.
Nutrition management of PKU with pegvaliase therapy: update of the web‑based PKU nutrition management guideline recommendations.
Orphanet Journal of Rare Diseases (2023) 18:155 doi.org/10.1186/s13023-023-02751-0
Updated: July 2023