V: LES NEPHROPATHIES HEREDITAIRES
Cours de Néphrologie Pédiatrique
MED 22 - Uro-Néphro
Etienne SOKAL
Retour Début du cours
-
Mutation portant sur la synthèse de la chaîne
alpha du collagène de type IV, composant de de la membrane basale
glomérulaire, mais également au niveau de l'oreille;interne
et de l'oeil (cristallin).
- Lié à l'X ==> hommes malades, femmes peu atteintes.
- Si pas d'antécédents familiaux, sans doute néo-mutation
spontanée.
-
Sclérose glomérulaire progressive
- Hématurie asymptomatique ou épisodes d'hématurie macroscopique
dès les premières années de vie
- Protéinurie plus tardive.
- Surdité progressive (10-25 ans)
- Déformation du cristallin
- Insuffisance rénale à l'âge adulte chez l'homme (20-40
ans)
Hyperoxalurie Primaire
- Déficit enzymatique hépatique (glyoxylate alanine transférase)
responsable de la formation excessive d'oxalate (voie métabolique alterne),
excrété par les urines. Les cristaux d'oxalate s'accumulent
dans le parenchyme rénal et provoquent uen néphrite tubulo interstitielle
chronique. Au stade d'insuffisance rénale, les cristaux se déposent
dans tout de l'organisme.
- Autosomial récessif (<==> consanguinité parentale)
- Symptômes apparaissent dans les premières années
- Polyurie, polydipsie, retard de croissance
- Lithiases urinaires ou néphrocalcinose diffuse
- Crises de coliques néphrétiques.
- Insuffisance rénale terminale avant 15 ans
- Accumulation ensuite généralisée d'oxalate: coeur,
rétine, os, artères...
- Diagnostic par dosage de l'enzyme sur le foie et recherche mutations
- Diagnostice anténatal possible si 2 allèles mutants identifiés
Traitement :
- Hyperhydratation, alcalinisation des urines (citrate de Na, 150 mg/kg).
Réduction calciurie
- Certaines formes (~ 50%) répondent au moins partiellement à
la pyridoxine, co-facteur de la GAT (activité résiduelle)
- Transplantation hépatique préemptive ou hépatique et
rénale si IR terminale
.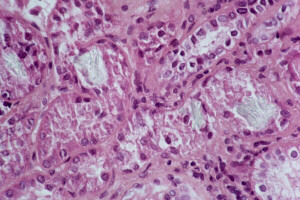 Cristaux d'oxalate dans
le parenchyme rénal
Cristaux d'oxalate dans
le parenchyme rénal
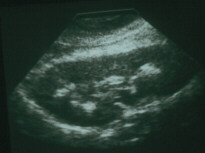 Ultrasons et AAB: précipitations
oxalates dans le rein
Ultrasons et AAB: précipitations
oxalates dans le rein 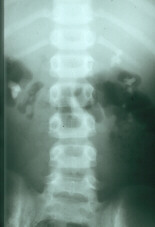
Acidose Tubulaire de de
Toni Debré Fanconi.
- Dès les premiers mois de vie
- Tubulopathie complexe. Néphrite interstitielle.
- Fuite urinaire de glucose, acides aminés, phosphore, potassium,calcium,ac
urique...
- Polyurie, Polydipsie
- Retard de croissance
- Déshydratation hyponatrémiques
- Acidose chonique tubulaire
- Rachitisme vitamino résistant
- Insuffisance rénale progressive dans l'enfance avant la puberté
Le syndrome de Toni Debré Fanconi a plusieurs étiologies dont::
- Néphronophtise
- Autosomique récessif
- Acidose tubulaire
- Dysplasie multikystique
- Début insidieux
- Polyurie,polydispise, retard de croissance
- Insuffisance rénale progressive
- Cystinose
- Autosomique récessif
- Cheveux blonds très clairs et pâleur des téguments (cfr
photo)
- Polyurie, Polydispise, Retard de croissance
- Acidose tubulaire majeure, polyurie, polydispise , rachitisme vitamino résistant,
protéinurie tubulaire, insuffisance rénale
- Hépatomégalie liée à l'accumulation de cystine
dans les cellules de Küpfer (maladie de surcharge). Déficit en
cystinosine, responsable du trnasport de cystine en dehors du lysosome
- Traitement par Cystéamine, qui favorise la sortie de cystine
du lysosome
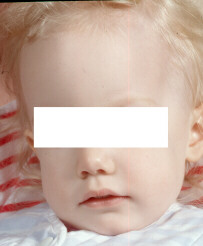 Cheveux blonds très
clairs et pâleur des téguments
Cheveux blonds très
clairs et pâleur des téguments 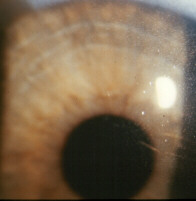 Cristaux cornée
Cristaux cornée
- Cystinurie:
Déficit différent de la cystinose: La cystinurie est liée
à un déficit héréditaire (autosomial récessif) du transport des
acides aminés dibasiques : cystine, ornithine, lysine et arginine. Ce déficit
entraine une élimination urinaire majorée et un trouble de l'absorption
intestinale de cystine. La lithiase cystinique est la seule manifestation clinique
de la cystinurie, cause principale de lithiase urinaire chez lenfant. La formation
de calculs est liée à la faible solubilité de la cystine
dans l'urine. Le défaut dabsorption digestive des acides aminés na pas de
conséquence clinique, car ces acides aminés sont absorbés dans
les oligopeptides qui les contiennent et qui sont normalement absorbés par le
tube digestif.
Traitement: Régime pauvre en méthionine (précurseur de
la cystine). Augmenter diurèse par hyperhydratation, alcanisationdes
urines (citrate de K/Na, 150 mg/kg/jr)
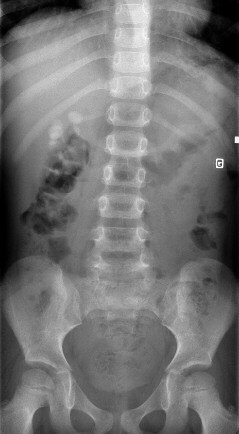 Lithiase rénale-
Cystinurie
Lithiase rénale-
Cystinurie
Site web spécialisé
Polykystose infantile (ARPKD= autosomial recessive
polykystic kidney disease)
- Affection autosomiale récessive
- Bilatéral
- Formes néonatales, néphromégalie
- Apparition dans l'enfance ou l'adolescence
- Déficit pouvoir de concentration, polyurie. HTA
- Insuffisance rénale terminale parfosi durant l'adolescence, parfois
tardive.
- Dilatation kystiques des tubes colecteurs
- Dédifférenciation cortico-médullaire
- Associée à la fibrose hépatique congénitale:
élargissement fibreux des espaces portes, parfosi hépatomégalie
majeure, hypertension portale pré-sinusoïdale
Mode de présentation possible par rupture de varices oesophagiennes.
Traitement
- Symptomatique de l'insuffisance rénale, HTA
- Ligature de varices, TIPS, shunt en général bien tolérés
au vu de la fonction hépatique normale
- Transplantation hépatique et rénale combinée parfois
nécessaire dès l'enfance