Quantum Transport in Carbon Nanotubes

Contacts: Jean-Christophe Charlier
Introduction
Single-wall carbon nanotubes (SWNTs) exhibit either a metallic or a semiconducting behavior depending on their helicity. Metallic tubes have mm long mean free paths and behave as long ballistic conductors. Conversely, the intrinsic properties of conducting multiwalled carbon nanotubes (MWNTs) are elusive. Indeed, reported ballistic, diffusive, or insulating experimental behaviors remain difficult to relate with the number and helicities of constitutive shells and the relevance of interlayer coupling. It is usually assumed that the outermost shell, in contact with metallic electrodes, determines the metallic or semiconducting character of the MWNTs.
Mesoscopic transport in chemically doped carbon nanotubes
In order to tailor the electronic properties of CNTs, the Fermi level can be tuned by chemical doping. Carbon nanotubes doped either with nitrogen or boron substitutions have been synthesized. When substituting a carbon atom, the boron atom (nitrogen) acts as an acceptor (donor) impurity in the nanotube. First-principle studies of the electronic structure and quantum transport in boron-doped (nitrogen) carbon nanotubes using the Landauer formalism have revealed the presence of localized states around the substituting impurity. These quasibounded states are responsible for an enhancement of the backscattering for energies below (above) the charge neutrality point.
Electronic quantum transport is investigated in boron- and nitrogen-doped carbon nanotubes (between 10 nm and 1 mm long), using tight-binding methods correlated to ab initio calculations. The orthogonal tight-binding approach (zone folding technique) takes into account only one orbital p per carbon atom and describes correctly the usual electronic properties of pristine carbon nanotubes (gaps, positions of Van Hove singularities, Fermi velocity, etc.). This present technique also accurately accounts for both effects of dopants, namely, charge transfer and elastic scattering. Generic transport properties such as conduction mechanisms, mean-free paths, and conductance scalings are derived for various concentration of randomly distributed boron and nitrogen dopants. Our calculations allow direct comparison with experiments and demonstrate that a small amount of dopants (<0.5%) can drastically modify the electronic transport properties of the tube, which is certainly a key effect feature for envisioning nanoelectronics.
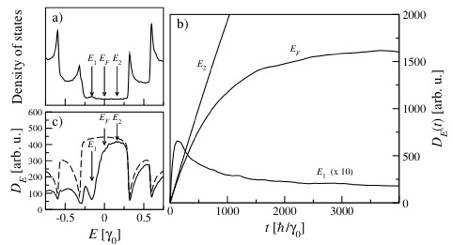
Electronic diffusion in a (0.1%) B-doped (10,10) CNT. (a) Density of states illustrating the characteristic acceptor peak located below the Fermi level (here EF=0). (b) Diffusivity DE(t) as a function of time for the three energies, indicated by arrows in (a). The difference of diffusion, according to the conduction regime is clear: ballistic (E2), diffusive (EF), and localized (E1, for which the coefficient is 10 times magnified). (c) The diffusivity DE plotted as a function of energy for the same B-doped CNT (solid line) and a pristine CNT (dashed line).
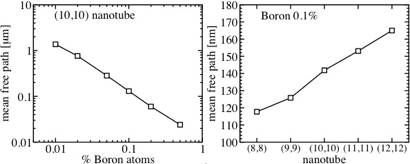
Scaling of the mean-free path le at the Fermi level, for a B-doped (n,n) nanotube. Left: in the case of a (10,10) nanotube with various boron concentrations, le behaves like the inverse of the doping rate. Right: for a fixed concentration of B atoms, le is roughly a linear function of the diameter.
Structural and electronic properties of carbon nanotubes decorated by physisorbed molecules
Aromatic compounds are known to interact weakly (physisorption) with graphite, and consequently also with the graphitic sidewalls of CNTs. This kind of noncovalent functionalization of CNTs with organic molecules does not significantly perturb the atomic structure of the CNT in contrast to its covalent counterpart. On the other hand, the presence of organic molecules on the sidewall of a CNT could modify its electronic and transport properties. Besides, a good knowledge of the CNTs reactivity and of the repercussion of adsorption are thus needed for their potential application as sensors.
The weak intermolecular forces between two sp2-like systems, including the van der Waals interactions, are often called p-stacking interactions as they originate from interacting p electrons of the two systems. These interactions are for example responsible for the interlayer bonding in graphite and are also present in the solubilization of CNTs in aromatic solvents. Though the concept of p electron is only valid for molecular systems presenting a planar symmetry, it can be extended, following the idea of Haddon and its p orbital axis vector (POAV), to nonplanar systems with tricoordinated carbon atoms.
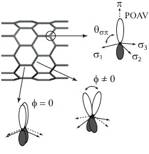
Schematic representation of a p orbital on a carbon atom, directed along the POAV and orthogonal to the three s orbitals. A schematic view illustrates the different misalignment angles f for the two inequivalent bonds of a (9,0) CNT.
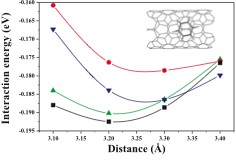
Calculated adsorption curves of a benzene molecule on a (8,2) CNT (see inset). The configurations with the molecule over different types of C–C bonds with a different value of f are considered: f =8.5° (squares), f =14.5° (down triangles), and f =23.0° (circles). The adsorption curve over a carbon atom (“stack” configuration) is also represented (up triangles).
Quantum transport in carbon nanotubes with random coverage of physisorbed aromatic molecules
Chemical sensing capabilities of carbon nanotubes-based devices appear very promising. Indeed, functional groups can specifically be attached to the carbon nanotube surface either by physisorption or by covalent bonding. Conventional covalent functionalization significantly perturbs the atomic structure of the CNTs and its corresponding electronic properties. On the other hand, the physisorption of organic molecules on the nanotube sidewalls is an example of non-covalent functionalization involving p-stacking interactions and corresponding to a weaker binding energy. The main interest in non-covalent functionalization stems from the negligible charge transfer involved within the p-stacking interactions. The induced scattering is thus expected to be low and molecular-dependent in opposition to the electrochemical covalent functionalization. Consequently, a good knowledge of the CNTs reactivity and of the impact of the physisorption on the quantum transport are needed to guarantee their potential application as chemical sensors.
The chemical sensitivity of electronic transport in carbon nanotubes under the physisorption of molecular species has been investigated using a tight-binding scheme, parametrized by first-principles calculations. Such a computational method enables to tackle the complex electronic properties of chemically grafted conducting nanotubes. The focus is made on the evaluation of the scattering strength, produced by a random coverage of p-conjugated hydrocarbon molecules (benzene (C6H6) and azulene (C10H8)), and acting on the wave packet propagation along the tube axis. Our calculations demonstrate that the impact of physisorption on the transport regime critically depends on the HOMO-LUMO gap of the attached molecules. In addition, the electronic mean free path exhibits a downscaling law with a lower dependence on the coverage density of grafted molecules than for conventional substitutional doping or homogeneous disorder.
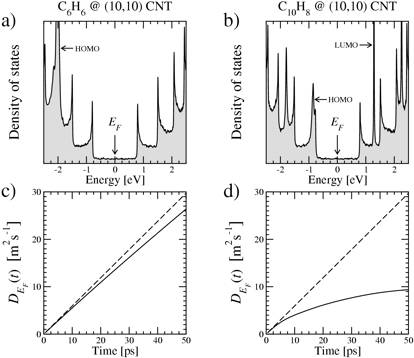
(a) DOS of the (10,10) CNT with a C6H6 density coverage of 16.3%. The HOMO molecular state is located by an arrow. (b) Same as in (a) but for a C10H8 density coverage of 11.5%. HOMO and LUMO levels are identified by arrows. (c) Time-dependent diffusion coefficient (at Fermi level) for the C6H6@CNT system, showing quasi-ballistic behavior (solid line). Ballistic conduction for pristine CNT case is also reported (dashed line). (d) Time-dependent diffusion coefficient for the C10H8@CNT structure, showing saturation at large times (diffusive regime).
Related publications
1) Mesoscopic transport in chemically doped carbon nanotubes
S. Latil, S. Roche, D. Mayou, and J.-C. Charlier
Physical Review Letters 92, 256805 (2004)
2) p-stacking interaction between carbon nanotubes and organic molecules
F . Tournus, S. Latil, M.I. Heggie, and J.-C. Charlier
Physical Review B 71, 165421 (2005)
3) Ab initio study of benzene adsorption on carbon nanotubes
F . Tournus, S. Latil, M. Heggie, and J.-C. Charlier
Physical Review B 72, 075431 (2005)
4) Electronic transport in carbon nanotubes with random coverage
of physisorbed molecules
S. Latil, S. Roche, and J.-C. Charlier
Nano Letters 5, 2216-2219 (2005)
Main collaborations
Dr. Sylvain Latil, Facultés Universitaires Notre-Dame de la Paix, Namur, BELGIUM
Dr. Stephan Roche, CEA-DSM/DRFMC/SPSMS, Grenoble, FRANCE
Dr. Florent Tournus, CEA, Grenoble, FRANCE
Main funds
Research Training Network, contract N° HPRN-CT-2000-00128, « COMELCAN : coupled
mechanical and electronic properties of carbon nanotubes based systems », 2000–2003.
Action de Recherche Concertée , « Interaction électron-vibration dans les nanostructures », 2001–2006.
Pôle d’Attraction Interuniversitaire P5/01, « Quantum size effects in nanostructured materials » (UCL-FUNDP-KUL-RUCA-UIA) 2002–2006.
European Network of Excellence (N° NMP4-CT-2004-500198), « NANOQUANTA : Nanoscale Quantum Simulations for Nanostructures an Advanced Materials » 2004–2008.