Also called the ornithine cycle,this cycle takes place in the periportal hepatocytes of the liver. It takes place partly in the cytoplasm and partly in the mitochondria to transform NH4 into urea (scheme). This cycle also generates H+ protons, which explains why metabolic alkalosis is common in case of hepatic failure..
The proper functioning of the cycle can be altered by an anomaly of one of the 6 enzymes (see specific sheets) that compose it, or of a transmembrane transporter (causing triple H syndrome).
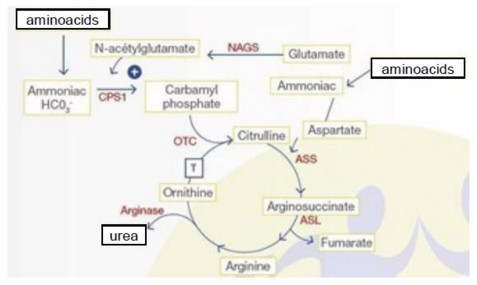
Physiologic reminder:
in the mitochondria,
- carbamyl phosphate synthetase I (CPS I, 2q35), produces carbamyl-phosphate from NH4+, ATP and CO2.. This reaction is done in two stages, each consuming one molecule of ATP. SPC I must be activated by N-acetylglutamate. This reaction is inhibited by valproate, which explains why valproate (Depakine) can be the cause of a decompensation of an hitherto paucisymptomatic abnormality of the urea cycle.
- N-acetylglutamate is synthesized by the combination of glutamate with acetyl-CoA by N-acetylglutamate synthetase (NAGS, 17q21.31)
- carbamyl phosphate combines with ornithine to give citrulline. This reaction is catalyzed by ornithine transcarbamylase (OTC, Xp21.1).
- citrulline is exported to the cytoplasm by the mitochondrial membrane ornithine and citrulline transporter (ORNT1, 13q14). ORNT1 deficiency causes triple H syndrome (hyperornithinemia - hyperammonemia - homocitrullinuria).
in the cytoplasm,
1) arginosuccinate synthetase (ASS, 9q34) combines citrulline, in the presence of ATP, with one aspartic acid molecule to produce arginosuccinate
2) arginosuccinate is converted to arginine and fumaric acid by arginosuccinate lyase (ASL, 7q11.2).
3) arginase 1 (ARG1, 6q23) catalyzes the hydrolysis of arginine into ornithine and urea by consuming one molecule of H2O. Ornithine is thus regenerated and can return to the mitochondria to bind to a new carbamyl-phosphate molecule with ORNT1.
Clinical signs are variable: metabolic coma, neurological coma (convulsions, focal signs), hepatic coma (hepatomegaly, cytolysis, hepatocellular failure) or neuropsychiatric coma (ataxia, behavioral disorders). They also vary according to the age of onset of the first symptoms:
- neonatal period: hyperammonemia with vomiting, lethargy or irritability;
- infantile period: anorexia, vomiting, developmental delay, hepatomegaly. A digestive cause for these problems is often searched for before making the diagnosis;
- childhood and adolescence: acute encephalopathy (Reye syndrome) during stress (fever, protein catabolism, postoperative period) or after valproate intake (Depakine), or progressive neurological signs (mental retardation, convulsions, ataxia). Corticosteroid therapy (increased protein catabolism) or an asparaginase-based chemotherapy may also be a triggering factor. Some patients experience a spontaneous distaste for meat or protein-rich meals.
- the postpartum is also a period at risk of decompensation because of the catabolic reaction induced by uterine involution.
Basic principle of treatment: prevention of hyperammonemia by a low-protein diet (the protein content is adapted according to age, taking growth into account and is reduced in case of infection or significant stress such as surgery to limit protein catabolism and NH4 formation. In addition, it is often necessary to add Na benzoate (0.25 g / kg / d), Na phenylbutyrate (0.25 g / kg / d) to stimulate alternative metabolic pathways and decrease the synthesis of NH4, or arginine HCl (0.1 to 0.2 g / kg / d) to promote the formation of urea via other routes.
Anesthetic implications:
- contact the team that usually has the child in charge
- shorten the duration of preoperative fasting: administer a glucose-rich solution as soon as the fasting period begins; in case of elective surgery, stop protein intake 24 to 48 hours before the procedure and compensate for caloric intake with glucose and lipids;
- empty the stomach to avoid protein intake through the digestive tract in case of surgery where blood can be swallowed (ENT, stomatology);
- special monitoring: NH4 (nl: < 50 ěmol/L or 20-80 ěg/dL), blood glucose;
- provide anesthesia that reduces the stress reaction: locoregional techniques, opioids, optimal postoperative analgesia; avoid dexamethasone which increases protein catabolism; careful use of antiemetics because they could mask the first signs of encephalopathy.
- add Na benzoate (0.25 g / kg / day) and Na phenylbutyrate (0.25 g / kg / d) which promote alternative metabolic pathways and reduce the synthesis of NH4, and arginine HCl (0.1 to 0.2 g / kg / d) to IV solutions; these solutions can cause hypokalemia
- in case of hyperammonemia > 3 x normal: glucose 10 or 20 % IV, and loading dose of Na benzoate (0.25g / kg) and Na phenylbutyrate (0.25g / kg), and depending on the specific deficiency: HCl arginine (0.2 g / kg) and carnitine (0.2 mg / kg / d) IV;
- in case of failure of the medical treatment: hemofiltration or peritoneal dialysis
References :
- Dobbelaere D, Mention K.
Déficits du cycle de l’urée in Maladies métaboliques héréditaires, éditeurs B Chabrol et P de Lonlay,
Progrčs en pédiatrie n°29, p139-147, Doin 2011.
- Del Rio C, Martin-Hernandez E, Ruiz A, Quijada-Fraille P, Rubio P.
Perioperative management of children with urea cycle disorders.
Pediatr Anesth 2020; 30:780-91.
Updated: August 2021