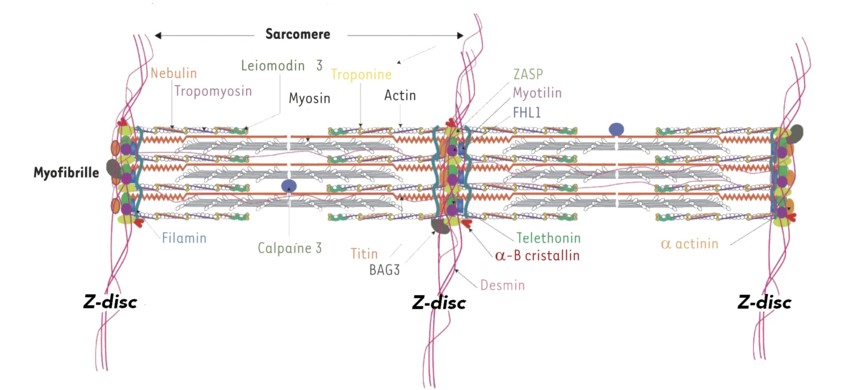
Group of muscular pathologies (skeletal and/or cardiac muscle) caused by a mutation of the titin gene (TTN (2q31.2). Titin or connectin is the largest protein in the human body, and is the most abundant protein in the striated muscle after myosin and actin. This elastic protein bound to the myosin filaments is the main constituent of the longitudinal filaments supporting the myofibrillar architecture. At the A-band level, titin has multiple binding sites with myosin, protein C and AMP deaminase. At the band I level, it is bound to calpain 3. At the Z-band level, its association with actin guarantees the stability of the myofibrillar assembly during the muscle contraction.
More than 127 mutations of the TNT gene, associated with at least 10 different phenotypes (myopathy, cardiomyopathy or both), depending on whether a skeletal and cardiac muscle isoform is affected, were described in 2020. These mutations can lead to dilated or hypertrophic cardiomyopathies, arrhythmogenic right ventricular dysplasia and a dozen of different phenotypes of myopathic phenotypes.
Cardiac involvement:
- sarcomeric hypertrophic cardiomyopathy (< 5 %), autosomal dominant transmission: asymmetric hypertrophy of the LV (usually of the interventricular septum), risk of sudden death (about 1 %/year)
- dilated cardiomyopathy (CMD1G)(15-25 %), autosomal dominant transmission: dilatation of the LV or biventricular dilatation associated with systolic dysfunction; progression to heart failure
- restrictive cardiomyopathy
- arrhythmogenic ventricular cardiomyopathy (dysplasia) (see this term)
- more rarely: non-compaction of the left ventricle (see this term), ASD, VSD
Muscle involvement (table): a distinction is made between
A. Forms with congenital or infantile onset: often antenatal onset (fetal immobility, arthrogryposis). In general, motor delay and non-selective muscle weakness, with a relatively diffuse involvement, but a predominantly proximal and axial involvement (paravertebral muscles, diaphragm), and a retractile component (retractions of the Achilles tendons, Rigid Spine) can be present. A primary myocardial involvement (dilated cardiomyopathy) is not always present.
- Multi-minicore myopathy (see this term): autosomal recessive transmission, associated with cardiac involvement (MmD with heart disease). It includes EOMFC (Early-onset Myopathy with Fatal Cardiomyopathy): image of congenital myopathy with cores, progressive dilated cardiomyopathy, death in adolescence and phenotype similar to the Emery-Dreifuss myopathy
- LGMDJ2 or LGMDR10 girdle dystrophy (see this term)
- arthrogryposis multiplex congenita (see this term) and non-compaction of the left ventricle.
B. Forms starting after 10 years of age (juvenile) or in adulthood: histology; dystrophy or myofibrillar images
- tibial muscular dystrophy type Udd (Finland) (TMD, acronym for Tibial Muscular Dystrophy)[MIM 188 840]: steppage gait
- HMERF (acronym for Hereditary Myopathy with Early Respiratory Failure or Edström myopathy): appearance of a restrictive syndrome in adolescence, tetraparesis. Normal CPK levels.
- Emery-Dreifuss-like myopathy: high CPK levels (10x), contractures, no cardiac involvement
- Emery-Dreifuss type titinopathy (see this term) autosomal recessive transmission
- autosomal recessive juvenile distal titinopathy
- myofibrillar myopathies
- necklace myopathy
At histologic examination, one can observe, depending on the phenotype: dystrophic lesions, lined vacuoles, cores, nuclear centralization, myofibrillar clusters, eosinophilic inclusions (necklace), a predominance of type 1 fibers, fibers of irregular size.
|
name |
trans
|
age of onset |
phenotype |
muscle biopsy |
|
tibial muscular
|
AD |
>35 years |
deficit of levator foot muscles |
moderate fibrosis lined vacuoles |
|
early onset tibial muscular dystrophy |
AR |
20 years |
deficit of levator foot muscles |
dystrophy |
|
LGMDJ2 or LGMDR10 limb girdle myopathy |
AR |
<20 years |
quickly progressive deficit of the pelvic and scapular girdles |
lined vacuoles |
|
hereditary myopathy with early respiratory involvement (HMERF) |
AD (rare AR) |
>20 years |
distal then proximo-distal involvement rapid evolution Early respiratory involvement |
cytoplasmic bodies Lined vacuoles |
|
Emery Dreifuss-like myopathy |
AR |
<10 years |
limb girdle deficit early onset of retractions Rigid Spine respiratory involvement |
cytoplasmic bodies lined vacuoles
absence of minicores |
|
early myopathy with lethal cardiac disease (EOMFC) |
AR |
neonatal |
moderate motor deficit ptosis, facial paresis pseudo-hypertrophy of the calves severe and early dilated cardiomyopathy rapid evolution. death < 20 years |
dystrophic aspect minicores |
|
multiminicores congenital myopathy with cardiomyopathy (MmD with heart disease) |
AR |
<20 years |
predominantly axial deficit diffuse retractions
|
minicores
star-shaped
|
|
'centronuclear'
|
AR |
<20 years |
moderate axial and proximal deficit facial paresis restrictive syndrome absence of cardiomyopathy |
central nuclei in most of the muscular fibers minicores |
|
proximal muscular dystrophy |
AR |
adulthood |
less severe proximal deficit than LGMD2J |
dystrophic aspect |
|
LGMD with cardiomyopathy |
AD |
adulthood |
severe cardiomyopathy and moderate LGMD |
minicores |
|
distal myopathy of the lower limbs with posterior predominance |
AD |
adulthood |
distal deficit with proximal evolution Involvement of the calves |
central nuclei minicores |
|
arthrogryposis |
AR |
prenatal |
contractures with or without cardiomyopathy |
|
Anesthetic implications:
echocardiography; check the CPK levels; treatment of heart failure; sometimes implanted automatic defibrillator.
In case of muscle involvement, consider the patient as at risk of rhabdomyolysis induced by the halogenated agents and succinylcholine.
References :
- Juntas-Morales R, Perrin A, Cossée M.
Corrélation phénotype-génotype dans les titinopathies.
Cahiers de Myologie 2020; 21: 16-20.
- Oates EC, Jones KJ, Donkervoort S, Charlton A et al.
Congenital titinopathy : comprehensive characterization and pathogenic insights.
Ann Neurol 2018 ; 83 : 1105-24
- Wacker J, Di Bernardo S, Lobrinus JA, et al.
Successful heart transplant in a child with congenital core myopathy and delayed-onset restrictive cardiomyopathy due to recessive mutations in the titin (TTN) gene.
Pediatric Transplantation 2023; e14561. doi:10.1111/
petr.14561
Updated: March 2024