One in 1,000 children presents a severe or profound deafness at birth: the cause can be genetic or environmental (CMV embryopathy, for example). Deafness can also appear later due either to a genetic factor (progressive deafness) or an environmental factor: meningitis, labyrinthitis, acoustic trauma...
ReminderĀ: humans have a hearing threshold of around 0 decibels (dB). Above this threshold, sounds with higher sound pressure levels are heard as louder noises. Sounds above 90 dB can lead to chronic hearing damage if people are exposed to them every day or all the time. Hearing becomes uncomfortable if the sound pressure level is above 110 dB (threshold of discomfort).
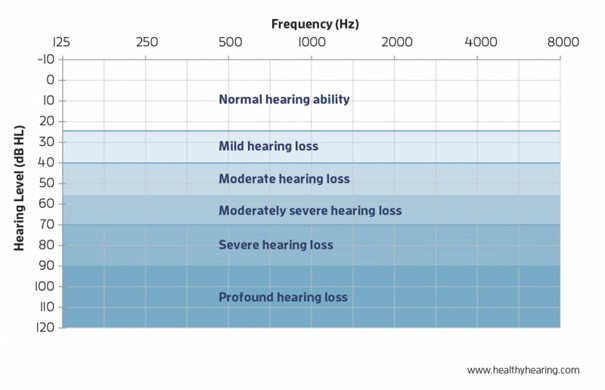
The severity of hearing loss is classified on the basis of the tonal loss:
- mild hearing loss: hearing loss of 20 to 40 decibels.
- moderate hearing loss: hearing loss of 41 to 60 decibels.
- severe hearing loss: hearing loss of 61 to 80 decibels.
- profound hearing loss or deafness: hearing loss of more than 81 decibels
Among the genetic causes, 10 % are part of a syndrome; the others are isolated or familial in the context of a monogenic disease
From a genetic point of view, the hearing losses are classified according to their mode of transmission: DFNB (autosomal recessive transmission), DFNA (autosomal dominant transmission), DFNX (X-linked transmission), DFNM (transmission by the mitochondrial genome) and DFNY (Y-linked transmission). The loci where mutations can occur are: DFNA 1 to 64. DNFB 1 to 95; DFNX 1 to 5; DFNY and 7 mitochondrial mutations. These genes code for a variety of proteins involved in hearing: proteins of the tight junctions (CDLNI4, TJP2), channels and transporters (SLC26A4, KCNQ4), enzymes, growth factors, ribosomal RNA, etc.
Typical features:
- nonsyndromic deafness: the most common are
- DFNB1 (the cause of 60 % of nonsyndromic deafnesses) and 30 % of sporadic deafnesses due to a mutation in the GJB2 gene coding for connexin 26; the predominant mutation is as common as that of cystic fibrosis; profound sensorineural deafness is present in 50 % of cases
- DFNB4 (5-6 %) due to a mutation in the SLC26A4 gene coding for pendrin: deafness of cochlear origin, that evolves in steps of sudden worsening)
- X-linked DFNX2 due to a mutation of the POU3F4 gene (Xq21.1) [MIM 304 400]: sensorineural hearing loss with a high transmission component; abnormalities on petrous temporal bone imaging; often a "geyser" ear due to communication between a cystic cochlea and the internal auditory canal (see LAMM, syndrome).
- syndromic deafness: deafness is included in hundreds of syndromes, such as
- Alport syndrome: progressive deafness with micro- and macroscopic hematuria due to a glomerulopathy. Progressive renal insufficiency.
- branchio-oto-renal (BOR) syndrome: association of anomalies of the branchial arches (clefts, fistulas), a transmission or sensorineural deafness, anomalies of the external ear (badly hemmed ears) and renal abnormalities (malformations, hypoplasia, agenesis, cysts).
- Jervell-Lange-Nielsen syndrome: long QT
- Pendred syndrome: due to a mutation in the same SLC26A4 gene as in DFNB4, it combines deafness of cochlear origin that often evolves in steps of sudden worsening, an euthyroid goiter (2th decade of life) due to a disorder of the organification of iodine. Morphological abnormalities of the petrous bones: dilation of the aqueduct and incomplete cochlea.
- Usher syndrome: association of deafness, disorders of balance and retinitis pigmentosa. Indication of early implantation of bilateral cochlear implants. The subtype I is accompanied by vestibular areflexia
- Waardenburg syndrome: association of deafness with pigmentation abnormalities (white hair, bicoloured iris, plaques of skin depigmentation). Sometimes associated with Hirchsprung's disease.
Anesthetic implications:
exclude the presence of a syndrome (clinical examination, ECG); presence of the parents for the induction and emergence of anesthesia (sign language, lip reading). Be careful with ototoxic antibiotics. The risk of anesthetic complications is mostly respiratory but is not different from the children of the same age. Specific procedures: hearing tests, cochlear implant surgery.
References :
- Blanchard M, Thierry B, Marlin S, Denoyelle F.
Aspects génétiques de la surdité.
Arch Pédiatr 2012 19;886-9. - Yeh JS, Mooney KL, Gingrich K, Kim JT, Lalwani AK.
Anesthetic complications in pediatric patients undergoing cochlear implantation.
The Laryngoscope 2011; 121; 2240-4.
- Gungor G, Bozkurt PS, Yener HM, Yilmaz YZ, Atas A, Yilat S, Hayir D.
Comparison of anesthetic agents on otoacoustic emission in childrenĀ: propofol vs ketamine.
Pediatr Anesth 2016Ā; 26Ā:752-8. - Darlong V, Khanna P, Baidya DK, Pandey CR et al.
Perioperative complications of cochlear implant surgery.
J Anesth 2015Ā; 29Ā: 126-30 - Madan HK, Kosare S.
Anesthesia for middle ear surgeries and cochlear implant.
Otorhinolaryngology Clinics: An International Journal 2015;7 :1-9. - Pairaudeau C, Mendonca C.
Anaesthesia for major middle ear surgery.
BJA Education 2019Ā; 19(5): 136e143
Updated: August 2024