[MIM 251 200, 603 802, 604 317, 604 321, 604 804, 608 393, 608 716, 612 703, 614 673, 614 852, 615 414, 616 080, 616 402, 616 486, 616 681 ]
Rare. Prevalence of 1/100,000 in Europe, most common in Pakistan. Disorder of neurological development characterized by a cranial circumference reduced at birth without significant abnormalities of brain architecture and with variable intellectual deficit.
Transmission is autosomal recessive. There are ten subtypes on basis of 9 identified genes: MCPH1, WDR62, CDK5RAP2, CEP152, ASPM, CENPJ, STIL, CEP63, CASC5, PHC1 and CEP135. Those mutations cause a reduction in the production of cortical neurons during embryonic neurogenesis. Mutations of the genes CDC6, CDT1, ORC1, ORC4, ORC6; WDR62 can cause more serious cortical abnormalities. Mutations of those genes can produce a Meier-Gorin syndrome (see this term). The MCPH and Seckel syndrome (see this term) belong to a same clinical spectrum, mutations (CENPJ, CEP152) resulting in one or another of the phenotypes.
Cranial circumference (CP) is less than 2 standard deviations (SD) at birth for age, sex and ethnicity. Microcephaly can be detected by the 32nd week of pregnancy. The cranial growth is then very slow, and the CP is reduced: under 3 SD before the age of 6 months, and usually between 4 and 12 SD in adults. There is a slight or moderate non-progressive intellectual deficit, without significant neurological deficit, and sometimes epilepsy (10%). A delay in early motor references and speech are common. Most of the patients are hyperactive.
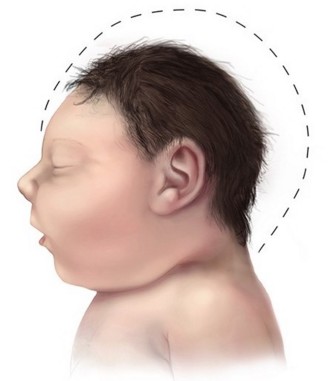
Anesthetic implications:
risk of difficult intubation if micrognathia; growth laryngotracheal is not affected by the microcephaly: choose the endotracheal tube according to age.
References :
- Nishida T, Mihara T, Horiki T, Ka K.
Anaesthesia and orphan diseaseĀ: primary autosomal recessive microcepaly-10 caused by a mutation of NF335 gene.
Eur J Anaesthesiol 2016; 33: 543-4
Updated: August 2016