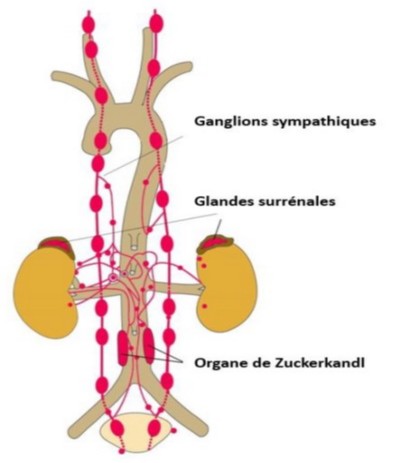
Tumoral of the chromaffin cells of the adrenal medulla or of the embryonic chromaffin extra-adrenal residues (bladder, Zuckerkandl organ in the pelvis) that secrete catecholamines in an uncontrolled manner. There are non-secreting (or rarely secreting) forms, called paragangliomas, that derive from the embryonic tissue that will produce the parasympathetic system and are generally located in the head and neck region.
Being a rare disorder (0.1 per million children), pheochromocytoma is responsible to be the cause of about 1 % of the arterial hypertension in children.
In summary:
- 10 % of pheochromocytomas are diagnosed before the age of 16 years;
- there is a clear male predominance (62 %) before puberty;
- the lesion affects both adrenals in 1/3 of the cases and is extra-adrenal in 18 % of the pediatric cases.
- the rate of recurrence after treatment is 12 % in children, mainly after conservative surgery, especially if the adrenal gland is involved.
It is traditionally taught that 10 % of pheochromocytomas are familial [MIM 171 300]. Progress of molecular genetics have shown that a genic or somatic mutation is actually found in 60 % of the pheochromocytomas discovered before the age of 18 years and 70 % of those diagnosed before the age of 10 years.
Several disorders and genetic syndromes are associated with a pheochromocytoma: they must be sought systematically.
They are sometimes grouped according to the molecular cause of the appearance of the tumor:
1) pseudohypoxic tumours with abnormal angiogenesis
- von Hippel Lindau syndrome (retinal angiomatosis, pancreatic and renal cysts, renal cell carcinoma, pheochromocytoma), autosomal dominant disease related to mutations of the tumor suppressor gene VHL (3p25.3) (acronym for von Hippel-Lindau); risk → 10-20 %
- familial paraganglioma (20 % risk): the tumor is generally located in the head and neck region and is possibly secreting;
there are several mutations of one of the genes coding for succinate-dehydrogenase (complex II of the mitochondrial respiratory chain):
- mutation of the SDHA gene (5p15) [MIM 614 165], familial paraganglioma type 5 (PGL5)
- mutation of the HBDS gene (1p36.13) [MIM 115 310], familial paraganglioma type 4 (PGL4)
- mutation of the SDHC gene (1q23.3) [MIM 605 373], familial paraganglioma type 3 (PGL3)
- mutation of the SDHD gene (11q23) [MIM 168,000], familial paraganglioma type 1 (PGL1)
- mutation of the SDHAF2 gene (11q13) [MIM 601,650], familial paraganglioma type 2 (PGL2)
Mutations of the FH gene (1q43) coding for fumarate hydratase can also produce a familial paraganglioma
- Sturge-Weber-Krabbe syndrome, following a somatic mutation of the GNAQ gene (9q21), associating capillary or cavernous hemangiomas, meningeal angiomas, convulsions, glaucoma, mental retardation and, sometimes, pheochromocytoma
- Pacak-Zhuang syndrome (mutation with gain of function of the EPAS1 gene (or HIF2A for hypoxia-inducible factor 2-alpha) (2p21) ombining early polycythemia, multiple paragangliomas (sometimes secreting), and digestive somatostatinoma (usually duodenal)
2) tumors involving activation of the MAPkinase pathway and neuroendocrine differentiation:
- neurofibromatosis type 1 (or Recklinghausen's disease), which is complicated by a pheochromocytoma in 1 % of cases; mutation of the NF1 gene (17q11.2)
- multiple endocrine neoplasia (MEN) 2A (Sipple syndrome), combining medullary thyroid carcinoma, hyperparathyroidism and pheochromocytoma in 50 % of cases (mutation of the proto-oncogene RET (10q11.2))
- MEN 2B (Gorlin syndrome), combining medullary thyroid carcinoma, pheochromocytoma (50 %), Marfan morphology, mucous neuromas and intestinal ganglioneuromas (mutation of the proto-oncogene RET (10q11.2))
- tuberous sclerosis of Bourneville, (TSC1 gene (9q34)), associating convulsions, intellectual retardation, facial angiofibromas, proliferating hamartomas pancorporeal and, in a small proportion of cases, pheochromocytoma;
- paraganglioma/familial pheochromocytoma caused by a mutation in the KIF1BP gene (1p36.22)[MIM 171,300], the TMEM127 gene (2q11.2) or a somatic mutation of the MAX gene (14q23.3)
Clinical presentation: highly variable; starts most often between the age of 6 and 14 years but it can occur at any age. The clinical presentation is usually dominated by headache and sweats in a context of nausea, fatigue, weight loss, abdominal pain with constipation, visual disturbances, pallor and anemia. At clinical examination, one notes a permanent systolo-diastolic hypertension, without hypertensive attacks but with a frequent significant effect on the myocardium (early signs of left ventricular hypertrophy) and on the retina (bilateral papilledema). The disease may be discovered as part of a hypertensive encephalopathy or brutal hemiplegia. In case of necrosis of the tumor (infarction or hemorrhage), the child may be in cardiogenic shock associated with the massive release of catecholamines. Sometimes it is a fortuitous discovery in presence of non-symptomatic cardiomyopathy. It can also happen that the problem, until there unknown, presents during general anesthesia as attacks of unexplained persistent or intermittent hypertension in a properly anesthetized child.
Some neuroblastomas are secreting catecholaminesand may present initially with a clinical picture similar to a pheochromocytoma.
Diagnosis:
- 24 h urinary excretion of metanephrine and normetanephrine;
- plasma levels of metanephrines, noradrenaline and epinephrine; it is useful to know what type of catecholamine is the most secreted: in case of systemic hypotension after resection of the tumor, an infusion of this catecholamine will be most effective to restore normotension.
- location of the tumor: MIBG scan to locate extra-adrenal tumors, TDM or, preferably, thoraco-abdominal MRI.
Anesthetic implications:
Different perioperative strategies can be applied to control BP : phenoxybenzamine, labetalol, a calcium channel blocker ... In case of dopamine secretion, a preoperative antihypertensive treatment with alpha-blockers (phenoxybenzamine or prazosin) may result in profound hypotension by in unmasking the vasodilating effects of dopamine on D1 receptors. Echocardiography (cardiomyopathy ?), ionogram, invasive monitoring. Many protocols exist to control BP during the procedure:
- induction: magnesium sulfate, prevent attacks of hyper- (intubation, insufflation peritoneal if laparoscopy) and hypotension
- before clamping the venous drainage of the tumor, hypertensive crises should be treated with an infusion of phentolamine (Regitine�, alpha blocker), a calcium channel blocker (nicardipine), or Na nitroprusside (vasodilator) associated with a short-acting beta-blocker (esmolol) in case of tachycardia
- risk of severe hyperthermia probably linked to a hypermetabolic crisis associated with an acute catecholaminergic discharge, that can be difficult to distinguish from a malignant hyperthermia crisis
- after the removal of the tumor: major hypotension: adrenaline or, more often, noradrenaline infusion
- risk of post-operative hypotension and hypoglycemia
References :
- Allen GC, Rosenberg H.
Phaeochromocytoma presenting as acute malignant hyperthermia: a diagnostic challenge.
Can J Anaesth 1990; 37: 593-5.
- Armstrong R, Sridhar M, Greenhalgh KL, Howell L, Jones C et al.
Pheochromocytoma in children.
Arch Dis Child 2008; 93: 899-904
- Hammond PJ, Murphy D, Carachi R, Davidson DF, McIntosh D.
Childhood phaeochromocytoma and paraganglioma: 100% incidence of genetic mutations and 100% survival.
J Pediatr Surg 2010; 45:383-6.
- Zerhouni
H, Kaddouri N, Abdelhak M, Benhmamouch N, Barahioui M.
Le phéochromocytome de l'enfant. À propos de deux cas.
Ann Urol 2002; 36:87-94.
- Tobias JD.
Preoperative blood pressure management of children with catecholamine-secreting tumors : time for a change.
Pediatr Anesth 2005; 15: 537-40.
- Seefelder C, Sparks JW, Chirnomas D, Diller L, Shamberger RC.
Perioperative management of a child with severe hypertension from a catecholamine secreting neuroblastoma.
Pediatr Anesth 2005; 15: 606-10.
- Hernandez MR, Shamberger RC, Seefelder C.
Catecholamine-secreting neuroblastoma in a 4-month-old infant: perioperative management.
J Clin Anesth 2009; 21: 54-6.
- Bryskin R, Weldon BC.
Dexmedetomidine and magnesium sulfate in the perioperative management of a child undergoing laparoscopic resection of bilateral pheochromocytomas.
J Clin Anesth 2010; 22:126-9.
- Bakan M, Kaya G, Cakmakkaya S, Tufanogullari B.
Anesthesia management with short acting agents for bilateral pheochromocytoma removal in a 12-year-old boy.
Pediatr Anesth 2006; 16:1184-8.
- Watanabe A, Shimoda T, Takeuchi M, Tachibana K, Kinouchi K.
Anesthetic management for a child with bilateral pheochromocytoma.
Masui 2010; 59:397-400.
- Dubey RK, Verma N, Pandey CK.
Anaesthetic management of a dopamine-secreting phaechromocytoma in multiple endocrine neoplasia 2B syndrome.
Indian J Anaesth 2014; 58: 217-9.
Updated: April 2022