[MIM 161 800, 256 030, 605 355]
(Nemaline rod myopathy, nemaline dystrophy )
Rare: 1/50.000 except for the Amish form where the prevalence is 1/500. In general, sporadic transmission (65%) but autosomal recessive and dominant forms exist. Ten genes are associated with this myopathy without any precise correlation between the genotype and the severity of the phenotype:
|
acronym |
gene |
locus |
|
protein |
MIM |
comment |
|
NEM1 |
TPM3 |
1q21.3 |
AD |
tropomyosin 3 |
609 284 |
|
|
NEM2 |
NEB |
2q23.3 |
AR |
nebulin |
256 030 |
|
|
NEM3 |
ACTA1 |
1q42.13 |
AD |
muscular
|
161 800 |
50% severe neonatal forms |
|
NEM4 |
TPM2 |
9p13.3 |
AD |
tropomyosin 2 |
609 285 |
|
|
NEM5 |
TNNT1 |
19q13.42 |
AR |
troponin T type 1 |
605 555 |
mostly Amish
|
|
NEM6 |
KBDT13 |
15q22.31 |
AD |
Kelch repeat abd BTP domain |
609 273 |
|
|
NEM7 |
CFL2 |
14q13.1 |
AR |
cofilin |
601 443 |
|
|
NEM8 |
KLH40 |
3p22.1 |
AR |
Kelch-like family member 40 |
615 348 |
|
|
NEM9 |
KLH41 |
2q31.1 |
AR |
Kelch-like family member 41 |
615 731 |
|
|
NEM10 |
LMOD3 |
3p14.1 |
AR |
leiomodin 3 |
616 165 |
|
|
NEM11 |
MYPN |
10q21.3 |
AR |
myopalladin |
617 336 |
|
At least 4 clinical presentations have been described:
- severe congenital form (ACTA-1, NEB, KLHL 40, LMOD3): massive muscle involvement with lack of spontaneous motricity requiring assisted ventilation and gavage feeding; death usually occurs in the 1st year of life.
- intermediate congenital form (NEB) (there is also a form known as the Amish nemaline myopathy: TNNT1): hypotonia and generalized muscle weakness appear after a few months of life; important developmental retardation: neither walking nor seating are acquired; scoliosis and muscle contractures with early respiratory failure.
- typical form (NEB, TMP2, KLHL41): onset at a variable age, diffuse muscle involvement with amyotrophy and marked muscle contractures, restrictive pulmonary syndrome, swallowing disorders and narrow facies with pro- or micrognathia, which can lead to intubation difficulties.
- form beginning in childhood or adolescence (TPM3): late onset of muscle weakness which is generally proximal
Other rarer forms:
- sporadic form starting in adulthood: quickly generalizing with death by cardiac involvement; approximately 50% of these late-onset forms are acquired and associated with a benign monoclonal IgG gammopathy (MGUS acronym for Monoclonal Gammapathy of Undetermined Significance ) and respond partly to corticotherapy with or without azathioprine, even after a bone marrow transplantation.
- asymptomatic form: rare, only found at muscle biopsy during familial screening.
- a mostly cardiac form in cases related to a mutation the ACTA1 gene: dilated cardiomyopathy, congestive heart failure, ventricular rhythm disorders.
The CPK level is little or not elevated. Death is caused by progressive respiratory failure often aggravated by aspiration pneumonitis secondary to bulbar impairment.
In some cases, a mutation in the RYR1 (19q13.2) or NEB-KBTBD3 gene leads to the appearance of central "cores" and this variant is called core-rod myopathy (see central core disease).
An ACTA1 gene mutation can also cause the zebra bodies myopathy that is considered as be a variant of rod myopathy. Another variant is the cap myopathy where there is a rearrangement of the thin muscular filaments: it presents like a typical form of rod myopathy but the histological picture shows a "cap" in periphery of the muscle fibers. It is associated with mutations of ACTA1, TPM3 or TPM2 gene.
A mutation of the MYO18B gene (22q12.1) [MIM 616 549] leads to the combinaison of a rod myopathy, facial dysporphism and Klippel-Feil syndrome (see this term).
The nemalin rod myopathy is due to an autosomal recessive transmission of the RYR3 gene (15q13-q14).
The very rare nemalin rod bodies myopathy [MIM 620 310] is due to an autosomal recessive transmission of mutations of the RYR3 gene (15q13-q14). The muscle biopsy shows wide variation in fibre size with type 1 fibre predominance and atrophy with abundant nemaline bodies located in perinuclear and subsarcolemmal areas, and within the cytoplasm.
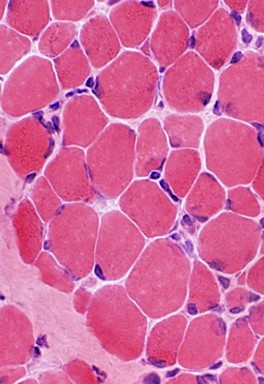
In the nemaline myopathy,rods are visible in optical or electron microscopy and correspond to cytoplasmic inclusions derived from proteins (actin, α-actinin) in the Z zone of the muscle fibers.
Anesthetic implications:
consider it in cases of delayed gait acquisition or rapid muscle fatigue on exercise, or in association with dysarthria, contractures or scoliosis. Cardiac ultrasound: cardiomyopathy? pulmonary hypertension secondary to chronic hypoxemia ? The patient is sometimes equiped with an automatic internal cardioverter defibrillator.
No case of malignant hyperthermia associated with nemaline myopathy has been published. However, in cases of RYR1 mutation or central cores, there is a risk of malignant hyperthermia: this precaution is prudently extended to all patients suffering from this genetic form of myopathy, whose precise genotype is unknown. As with all myopathies, succinylcholine should be avoided, but halogenated agents have been used without causing rhabdomyolysis. Risk of difficult intubation: early involvement of the face and neck muscles. Nocturnal ventilation in cases of severe respiratory insufficiency. Amyotrophy and swallowing disorders.
References :
- Shenkman Z, Sheffer O, Erez I, Litmanovic I, Jedeikin R. Spinal anesthesia for gastrostomy in an infant with nemaline myopathy. Anesth Analg 2000; 91: 858-9.
- Heard SO, Kaplan RF. Neuromuscular blockade in a patient with nemaline myopathy. Anesthesiology 1983; 59: 588-90.
- Klingler W, Rueffert H, Lehmann-Horn F, Girard T, Hopkins PM. Core myopathies and risk of malignant hyperthermia. Anesth Analg 2009; 109: 1167-73.
- Deconinck N, Laterre EC, Van den bergh PYK.
Adult-onset nemaline myopathy and monoclonal gammapathy: a case report.
Acta Neurol Belg 2000; 100: 34-40. - Ryan MM, Schnell C, Trsickland CD, Shield LK et al.
Nemaline myopathy: a clinical study of 143 cases.
Ann Neurol 2001; 50: 312-20. - Desaegher J, Vanmarsenille J-M, Vekemans M-C, Van den Bergh PYK.
Myopathie némaline sporadique à début tardif avec gammapathie monnoclonale : traitement par cellules souches hématopoïétiques.
Louvain med 2014 ; 133 : 522-6. - Raveau T, Lassalle V, Dubourg O, Legout A, Tirot P.
Révélation d’une myopathie à bâtonnets de l’adulte par une insuffisance respiratoire aiguë après chirurgie ambulatoire de la cataracte.
Ann Fr Anesth Réanim 2012 ; 31 : 638-40. - Brislin RP, Theroux MC.
Core myopathies and malignant hyperthermia susceptibility : a review.
Pediatr Anesth 2013 ; 23 : 834-41. - Christiaens F, Van den Bergh PYK.
Les myopathies congénitales.
Louvain Med 2014 ; 133 : 478-90. - North K, Wang C, Clarke N, Jungbluth H, Vainzof M, Dowling J et al.
Approach to the diagnosis of congenital myopathies.
Neuromuscul Disord 2014 ; 24 : 97-116. - Tran NH, Chhibber A.
Anesthetic management of a pediatric patient with NEB1-genotype nemaline rod myopathy for cleft palate repair.
Pediatr Anesth Crit Care J 2016; 4: 78-82 - Tran NH, Smith D.
Anesthetic consideration for patients with nemaline rod myopathy: a literature review.
Pediatr Anesth Crit Care J 2017; 5: 31-9 - Oliveira M, Fernandes AL.
Using sevoflurane in a pediatric patient with nemaline rod myopathy.
Pediatr Anesth 2018; 18: 749-50 - Oliveira M, Fernandes AL.
Using sevoflurane in a pediatric patient with nemaline rod myopathy.
Pediatr Anesth 2018; 18: 749-50 - Benarroch L, Bonne G, Rivier F, Hamroun D.
The 2020 version of the gene table of neuromuscular disorders. Neuromusc Dis 2019Ā; 29Ā: 980-1018 ou http://www.musclegenetable.fr. - Jung H, Kim H, Lee SW.
Anesthetic management for surgery in a nemaline myopathy patient with difficult airway: a CARE-compliant case report.
Medicine 2023;102:46(e36174). doi.org/10.1097/MD.0000000000036174
Updated: July 2024