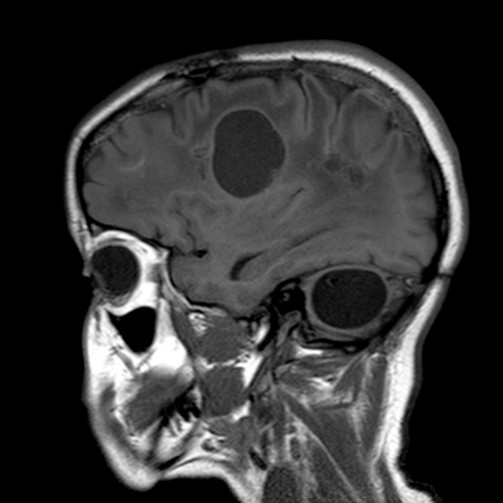
Very rare. Autosomal recessive transmission of a mutation of the SNORD118 gene (17p13.1). Variable onset, from childhood to adulthood. Progressive development of leucoencephalopathy with cerebral calcifications and formation of intracerebral cysts leading to cognitive decline, dyskinesia, spasticity, sometimes convulsions or hydrocephalus. The cause seems to be a diffuse microangiopathy of the cerebral vessels.
The clinical presentation is similar to the cerebroretinal microangiopathy syndrome with calcifications and cysts (CRMCC) [MIM 612 199] or to the Coats plus syndrome (see this term) caused by a mutation of the CTC1 gene but where osteopenia, a significant risk of digestive hemorrhage, anemia with thrombopenia and abnormalities of the skin and dander are observed.
Anesthetic implications:
neurological disorders, risk of intracranial hypertension (a case of neuroleptic malignant syndrome or serotoninergic syndrome after the administration of metoclopramide has been reported)
References :
- Shtaya A, Elmslie F, Crow Y, Hettige S.
Leukoencephalopathy, intracranial calcifications, cysts, and SNORD188 mutation (Labrune syndrome) with obstructive hydrocephalus.
World Neurosurg 2019Ā; 125Ā:271-2.
- Holland EL, Sanetto RP, Knipper EK.
Hypermetabolic syndrome and dyskinesia after neurologic surgery for Labrune syndromeĀ: a case report.
A&A Practice 2020Ā; 14Ā: e01212.
Updated: May 2020