(MIM 213 300, 610 688, 612 291, 614 173)
(Joubert syndrome type A, Joubert-Boltshauser Syndrome )
Rare: incidence estimated at 1/80,000 and 1/100,000. Autosomal recessive transmission, sometimes X-linked. Heterogeneous genetics: 10 genes have been identified including JBTS1/INPP5E (9q34), JBTS2/TMEM216 (11p12-q13), JBTS3/AHI1 (6q23), JBTS4/NPHP1 (2q13), JBTS5/CEP290 (12q21), JBTS6/TMEM67 (8q22), JBTS7/RPGRIP1L (16q12), JBTS8/ARL13B (3p12.3-q12.3), JBTS9/CC2D2A (4p15), JBTS10/OFD1.
These are actually mutations of the primitive cilium: it is thus a 'ciliopathy '.
The principal element is a malformation of the brainstem associated with hypoplasia or agenesis of the cerebellar vermis: at MRI, a typical "molar tooth" picture in axial section at the ponto-mesencephalic junction, caused by the association of hypoplasia of the vermis and malformations of the brainstem.
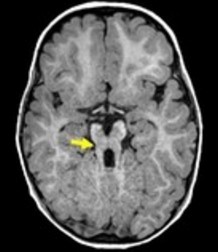
Clinical presentation :
- in the neonatal periode: irregular breathing (alternating periods of apnea and tachypnea during awakening and sleep) that improves gradually, and abnormal eye movements (nystagmus, lack of eye-tracking) associated with a retinal dystrophy
- later, hypotonia and cerebellar ataxia
- very variable, but often severe mental retardation
- various cerebral malformations: hydrocephaly, anomalies of the posterior fossa, anomalies of the corpus callosum, absence of hypophysis, polymicrogyria
- scoliosis
- sensitivity to the respiratory depressant effects of all general anesthetics, including N2O
The facies is characteristic: large head with a prominent forehead, epicanthus, high rounded eyebrows, upturned nose, open mouth with protrusion of the tongue which shows rhythmic movements, low set ears.
According to the associated malformations, 6 different phenotypes are described:
- JS or pure Joubert (or type A) syndrome: [MIM 213 300] no other associated malformation
- JS - O: with eye problem (or type B) (AHT1 gene in 20 % of cases): retinal dystrophy due to progressive degeneration of photoreceptor cells (sometimes Leber amaurosis)
- JS - N (NPHP1, RPGRIPIL genes) there is a renal pathology (nephronophthisis, cystic renal dysplasia) that causes progressive renal insufficiency.
- JS-gold (or cerebello-oculo-renal syndrome) (CEP290in 50 % of cases): with retinal dysplasia (sometimes Leber amaurosis)and nephronophthisis (Senior-Loken syndrome) [MIM 266 900] and/or cystic renal dysplasia (Dekaban-Arima syndrome) [MIM 243 910]
- JS - H: with congenital hepatic fibrosis (TMEM67 gene in 80 % of cases), which can progress to cirrhosis with portal hypertension; in case of association with a coloboma, it is known as COACH syndrome (MIM 216 360]
- JS - OFD (with an oro-facio-digital defect) (or oro-facio-digital type VI or Varadi Papp syndrome) [MIM 277 170]: there is a postaxial polydactyly, a bifid tongue or a tongue lobulated by hamartomas, occasionally a cleft palate; sometimes: absence of the pituitary gland, hypothalamic hamartoma or another brain tumor.
The various forms of Joubert syndrome may be associated with Hirschprung’s disease.
Anesthetic implications:
in the newborn: respiratory problems; later�: check the kidney and liver function; mental retardation; sometimes epilepsy (if other brain malformations are associated); increased susceptibility to respiratory effects of morphine: indication for remifentanil if an opioid is necessary? To provide sedation, dexmedetomidine seems to be ideal because it does not affect the control of breathing.
References :
- Habre W, Sims C, D'Souza M.
Anaesthetic management of children with Joubert syndrome.
Paediatr Anaesth 1997; 7: 251-3. - Vodopich D, Gordon G.
Anesthetic management in Joubert syndrome.
Paediatr Anaesth 2004; 14: 871-3. - Platis CM, Kachko L, Trabikin E et al.
Postoperative complications in Joubert syndrome.
Pediatr Anesth 2006 ; 16 : 799-800. - Brancati F, Dallapiccola B, Valente EM.
Joubert syndrome and related disorders.
Orphanet J Rare Diseases 2010; 5: 20. - Galante D, Meola S, Cinnella G, Dambrosio M.
Regional caudal blockade in a pediatric patients affected by the Joubert syndrome.
Acta Anaesth Scand 2009; 53: 693-4. - Brancati F, Dallapiccola B, Valente EM.
Joubert syndrome and related disorders.
Orphanet J Rare Diseases 2010; 5: 20 - Nagi S, Brahim I, Hammami N et al.
Signe de la “dent molaire”: aspect caractéristique en IRM du syndrome de Joubert.
Arch Pédiatr 2012 ; 19 : 74-6. - Buntenbroich S, Dullenkopf A.
Total intravenous anesthesia in a patient with Joubert-Boltshauser syndrome.
Pediatr Anesth 2013; 23: 204-5. - Bhaskar P, John J, Sivamurthy SK, Lone RA, Tysarowski PA et al.
Anesthetic management of an infant with Joubert syndrome for cardiac surgery.
J Clin Anesth 2013; 25: 488-90. - Sriganesh K, Vinay B, Jena S, Sughir V, Saini J, Rao GSU.
Anesthetic management of patients with Joubert syndrome: a retrospective analysis of a single-institutional case series.
Pediatr Anesth 2014;24: 1180-4 - Sarma P, Bindu PS, Dwarakanath S, Somanna S.
Association of oral-facial-digital syndrome type VI (Varadi-Papp syndrome) with optochiasmatic pilocytic astrocytoma.
Childs Nerv Syst 2015; 31: 789-92.
Updated: March 2020