(microdeletion 16p13.2, HAFOUS, syndrome of deafness-skeletal dysplasia-coarse facial features with thick lips)
Extremely rare. De novo mutation or autosomal dominant transmission of a mutation of the USP7 gene or a microdeletion in 16p13.2 that includes the USP7 gene. This gene codes for a protein regulating intracellular protein recycling and DNA repair. It presents features similar to Prader-Willi syndrome.
Association of:
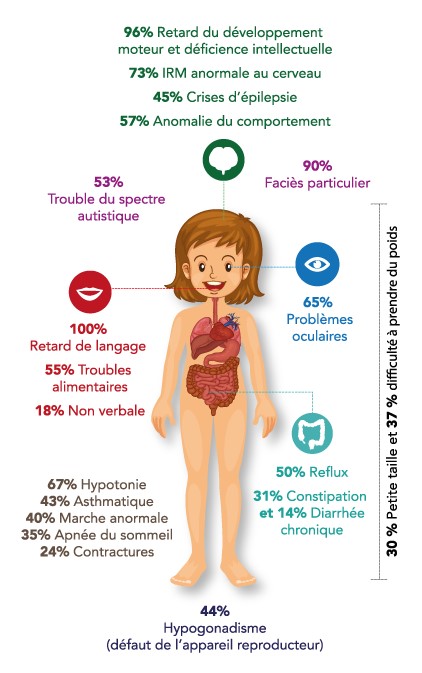
- delayed intellectual development of varying severity: delayed or non-acquisition of language, hearing disorders
- behavioral abnormalities: skin scratching, autistic-type disorders, hyperactivity, temper tantrums, aggressive or friendly behavior
- facial dysmorphism: thick lips, coarse features, etc.
But also :
- muscular hypotonia (79 %), with delayed motor development
- feeding difficulties in early childhood (56 %), with gastro-oesophageal reflux,
- abnormal pain threshold (76 %): either higher or lower than the general population
- early convulsions (40 %),
- sleep disorders (47 %), sleep apnea (29 %),
- tendency towards overweight and obesity due to hyperphagia (39 %)
- intestinal transit disorders
- hypogonadism (micropenis) (23 %),
- small, stubby hands and feet (30-40 %)
- abnormal gait
- eye problems
- kyphoscoliosis (30 %)
- body temperature instability (41 %)
Medical imaging: various abnormalities, especially in the white matter; hypoplasia of the corpus callosum
source: https://www.usp7.fr/
Anesthetic implications:
epilepsy, behavioral disorders, gastroesophageal reflux, pain threshold abnormalities, risk of hypo- or hyperthermia
References :
- Wimmer MC, Brennenstuhl H, Hirsch S, et al.
Hao-Fountain syndrome: 32 novel patients reveal new insights into the clinical spectrum.
Clinical Genetics 2024;105 :499‐509. doi:10.1111/cge.14480
Updated: June 2024