Prevalence is estimated at 1/400,000 newborns.
Two subtypes:
- transient neonatal diabetes [MIM 601 410]: very early onset (first days) with significant hyperglycemia, often without ketoacidosis; insulin needs are low and gradually decrease but the reappearance of diabetes in childhood or adolescence is frequent. The majority of the cases is due to an abnormality in chromosome 6
- final or permanent neonatal diabetes [MIM 606 176]: less frequent than the transient form, it is generally associated with a mutation or a syndrome; 30-50% of cases are due to a mutation of the gene coding for the potassium channel in the ß cell .
Genetic aspects (DMPN = Diabetus Mellitus Permanent Neonatal):
- PLAGL1 or HYMAI gene (6q24): usually, only the paternal allele is expressed. Diabetes appears in case of parental paternal disomy of chromosome 6 (41%), of duplication (6q24) of the paternal allele (29%) or hypomethylation of the maternal allele; presence of neonatal macroglossia, umbilical hernia.
- DEND syndrome (acronym for Developmental delay, Epilepsy, and Neonatal Diabetes): mutation of the genes KCNJ11 (potassium channel J11 [MIM 618 856, or DMPN 2] and ABCC8 (ATP-binding cassette transporter subfamily Cmember 8) in 11p15.1 [MIM 618 857 or DMPN 3]: autosomal dominant transmission; these mutations result in a defect of closure of the Kir6.2 or SUR1 potassium channel that allows the release of insulin. Sulfonylureas (tolbutamide) close this channel and allow insulin therapy to be stopped. In addition to permanent neonatal diabetes, presence of psychomotor delay of varying importance, epilepsy often resistant to treatment it is often accompanied by hypotonia. A less severe, intermediate form, known as iDEND, has been described.
- INS (insulin) (11p15.5) gene [MIM 618 583 or DMPN 4]: mutation causing a defect in the synthesis of insulin
- GCK gene coding for glucokinase (7p13) (or DMPN 1) regulating the metabolism of glucose in the β cell, and thus the amount of the released insulin; homozygous mutations (very rare) cause a complete deficiency while heterozygous mutations cause a mild intolerance to glucose and a MODY 2 (see this term)
- agenesis or isolated hypoplasia of the pancreas: IPF1A (10p12.2) [MIM 615 935], PDX1 (13q12.2) [MIM 260 370] or GATA6 (18q11.2) [MIM 601 656] genes
- FOXP3 (forkhead homeobox P3) (Xp11.23) gene coding for scurfine: X-linked syndrome IPEX (acronym for Immune dysregulation Polyendocrinopathy, Enteropathy, X-linked) that combines exfoliative dermatitis, refractory diarrhea with villous atrophy, hemolytic anemia, thrombocytopenia, glomerulonephritis, and autoimmune thyroiditis. Presence of antibodies against the glutamic acid decarboxylase (GAD)
- mitochondrial disease
- Berardinelli-Seip syndrome: insulin resistance, see this term
- Wolcott-Rallison syndrome (SLC19A2 gene): see this term
- hypoplasia / agenesis of the pancreas with congenital heart disease [MIM 600 001]: GATA6 gene (18q11.2)
- Hypoplasia / agenesis of the pancreas and cerebellar hypoplasia [MIM 609 069]: PTF1A gene (10p12.2)
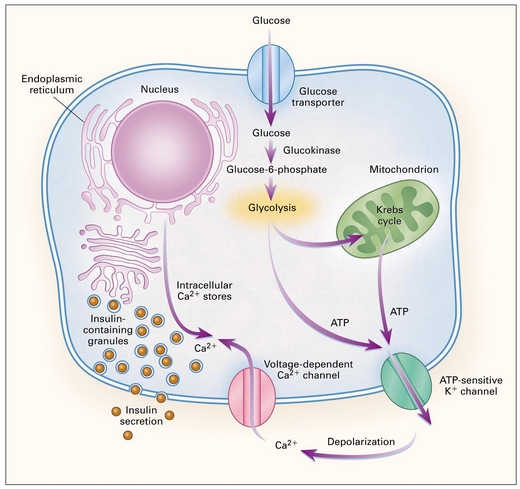
insulin secretion by ß cell
Anesthetic implications:
close monitoring of glycemia
References :
- Polak M, Busiah K, Cavé H.
Diabčte néonatal : maladie de l’empreinte génétique, mais pas seulement!
Métabolismes, Hormones, Diabčtes et Nutrition ; 2006 ; X, 21-8.
- Yafi M.
A case of neonatal diabetes: presentation, diagnosis and management.
Austin J Pediatr 2014; 1: 1004
- Kamoun T, Chabchoub I, Ben Ameur S, Kmiha S et al.
Transient neonatal diabetes mellitus and activating mutation in the KCNJ11 gene in two siblings.
Arch Pédiatr 2017; 24: 453-6
Updated: November 2022