(deficit in mitochondrial acetyl-CoA thiolase, deficiency in T2, alpha-methylaceto-acetic aciduria, MATD)
Extremely rare. Mitochondrial cytopathy (see this topic) due to the autosomal recessive transmission of a mutation in the ACAT1 gene (11q22.3). There are 6 intracellular thiolases (cytosol, peroxisomes, mitochondria): this deficiency concerns the mitochondrial thiolase and is therefore also called: mitochondrial acetyl-CoA thiolase deficiency. This enzyme is involved in the metabolism of ketone bodies and isoleucine.
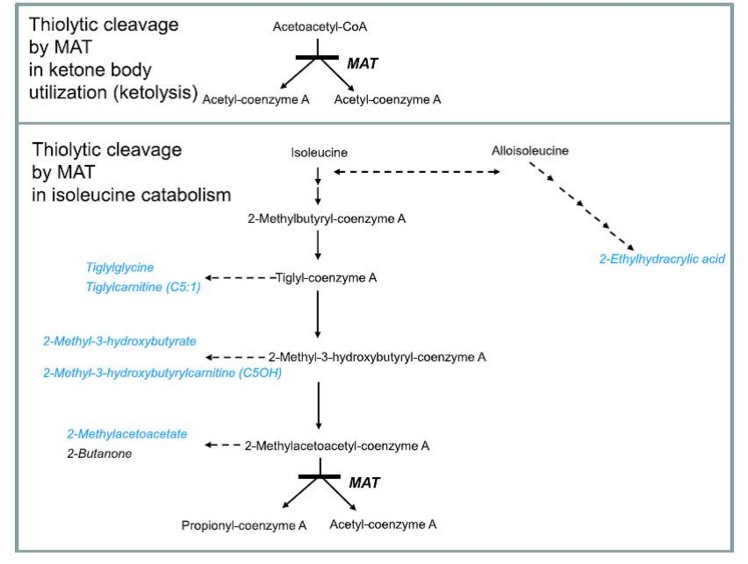
Onset in childhood (before 10 years of age, rarely in the neonatal period: median age 12 months) with episodes of severe cetoacidosis with or without hypoglycemia, sometimes complicated by coma, generally related to fasting or intercurrent infection, more rarely with a progressive loss of the acquired intellectual and motor skills.
M.R.I.: lesions of the basal ganglia.
Treatment: low protein diet (< 2g/kg/j), administration of IV glucose when fasting is required, carnitine supplements.
The prognosis is good if the diagnosis is early and the treatment well adhered to.
Anesthetic implications:
see mitochondrial cytopathies
References :
- Pandey R, Singh PM, Garg R, Darlong V, Punj J.
Perioperative concerns in a beta-ketothiolase-deficient child.
J Anesth 2015 ; 29 : 647.
- Grünert SC, Sass JO.
2-methylacetoacetyl-coenzyme A thiolase (beta-ketothiolase) deficiency : one disease, two pathways.
Orphanet J Rare Diseases 2020 ; 15 : 106
Updated: May 2020