[MIM 116 860, 603 284, 603 285]
(Hereditary cerebral cavernous hemangioma)
Prevalence: 1-5/10,000. Generally sporadic but 20 % of cases are hereditary: in these cases, there is an autosomal dominant transmission of a mutation of one of the CCM1 or KRIT1 (7q21.2), CCM2 (chr 7), or CCM3 (PDCD 10 on chr 3) genes.
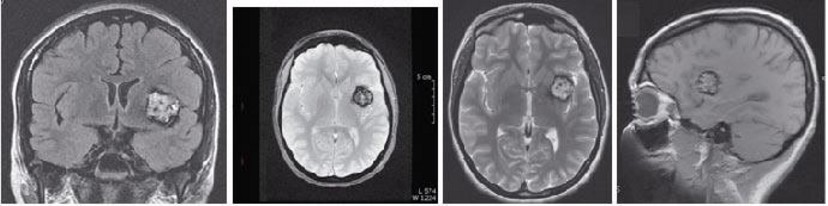
This vascular malformation consists in benign, highly proliferative lesions involving aberrant localized growth of capillary endothelium. They not demonstrable by arteriography; hence they are referred to as angiographically silent. These vascular capillary clusters are mainly located in the brain. The cerebral parenchyma surrounding these vessels is strictly normal. The size of these vessels varies from 1 mm to several cm. The number and the size of those vessels increase over time. In case of sporadic form, the cavernoma is generally unique. In familial forms, multiple cavernomas can coexist, and flat cutaneous angiomas are sometimes associated.
Hereditary cerebral cavernomas have been described in young children, but the majority of cavernomas appear between 20 and 50 years of age. The malformation is asymptomatic in 90 % of the cases.
The possible signs in the 10 % of symptomatic cases are:
- epilepsy (40-70 %)
- localized neurological deficit (35-50 %)
- headache (10-30 %) with incidental morning vomiting in case of intracranial hypertension
- cerebral hemorrhage.
The diagnosis is based on medical imaging (MRI).
Treatment: neurosurgery, gammaknife
Anesthetic implications:
according to the clinical presentation : seizures, cerebral hemorrhage, intracerebral vascular surgery
References:
- Reix G, Stoven C, Darcel I et al.
Cavernome cérébral familial révélé par une épilepsie chez une fillette de 10 ans.
Arch Pédiatr 2009 ; 16 : 1337-40.
Updated : November 2019