Incidence of 5-6/100.000 live births, up to 32/100,000 in French Polynesia. The pathogenesis is poorly understood and probably not unequivocal: anomaly of the development of the bile canaliculi, presence of abnormal bile acids, perinatal viral infection (respiratory syncitial virus, cytomegalovirus, Ebstein-Barr virus, reovirus, rotavirus...), genetic factors or autoimmune deficit.
Different forms:
- an embryonic or malformative form (10-20 %), where the atresia is associated with polysplenia and/or situs inversus with absence of retro-hepatic vena cava and hypoplasia of the portal vein, sometimes known as BASM syndrome (see this term).
- a perinatal form caused by infection (viral ?), metabolic disorder or a malformation limited to the hilar plate
Clinical presentation: neonatal jaundice due to conjugate bilirubin, with discolored stools ("putty") and dark urine. A hepatomegaly is present, often with splenomegaly. Jaundice gets worse alongside the appearance of a progressive biliary cirrhosis. The occurrence of ascites and signs of portal hypertension is usually delayed, after the 6th month of life.
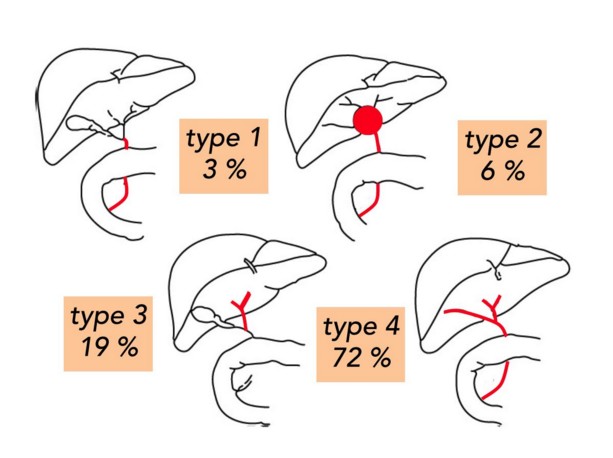
There are 4 types of the biliary atresia: type 1 (3%): common bile duct atresia; type 2 (6%): hilar cyst in communication with dystrophic biliary intra-hepatic tracks; type 3 (19%): the gallbladder, cystic and common bile duct are permeable; type 4 (72 %): complete atresia of the extra-hepatic bile duct
Some teams advocate a percutaneous needle biopsy or a percutaneous cholangiogram to confirm the diagnosis and avoid an useless laparotomy in a newborn with neonatal hepatitis, a metabolic disease, Alagille syndrome or progressive familial intrahepatic cholestasis (PFIC). The usual treatment is the Kasai or porto-enterostomy operation to drain bile in a Roux-en-Y bowel loop and try to slow down the evolution to cirrhosis. Other procedures can be applied: in type 1: cholecysto-enterostomy or hepatico-enterostomy; in type 2: kysto-enterostomy after checking by cholangiogram that the hilar cyst effectively communicates with dystrophic intrahepatic biliary ducts; in type 3: hepatoporto-cholecystostomy.
The prognosis of Kasai intervention is best if the child is operated before the age of 2 months and if the surgeon has a large experience of the intervention. More than 50 % of children develop chronic liver failure the only effective treatment of which is liver transplantation. If successful, the long-term prognosis is good: the 10-year survival rate is > 80 %, close sometimes to 95 %.
Even if the biliary drainage is effective, the fibrosis already present at the time of Kasai procedure continues to slowly evolve towards cirrhosis. This development is accelerated in case of cholangitis. In this case, the child will present repeated cholangitis and/or progressive portal hypertension with ascites and esophageal varices, leading to a gradual deterioration of liver function and stunted growth. It also happens that these children develop a hepatopulmonary syndrome or a portopulmonary hypertension (see these terms)
Anesthetic implications:
- neonatal period: check liver function; it is generally normal at the time of Kasai procedure; and the association of general anesthesia with thoracic epidural analgesia gives excellent results.
- later: check the liver function and the hemostasis; risk of portal hypertension; risk of low level of blood platelets in case of splenomegaly: malnutrition and osteopenia are frequent if ADEK vitamins and nutritional supplements have not been given
- echocardiography
ReferencesĀ:
- Chardot C, Carton M, Spire-Bendelac N, Le Pommelet C, Golmard JL, Auvert B.
Epidemiology of biliary atresia in France: a national study 1986-96.
J Hepatol 1999; 31:1006-13.
- Kelly DA, Davenport M.
Current management of biliary atresia.
Arch Dis Child 2007; 92:1132-5.
- Khalil BA, Perera MT, Mirza DF.
Clinical practice: management of biliary atresia.
Eur J Pediatr 2010; 169:395-402.
- Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G et al.
The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study.
J Pediatr 2006; 149:393-400.
Updated: February 2021