Heterogeneous group of degenerative eye diseases caused by mutations (> 250 identified) of the BEST1 gene (11q12). These mutations lead to highly variable clinical presentations, posing diagnostic and prognostic problems. It is possible that other genetic factors interacting with the BEST1 protein and environmental factors influence the phenotypic presentation of these mutations. A decrease in the Arden ratio on electro-oculography is characteristic of all these pathologies. This is the ratio of the light peak (LP) to the dark depression (DT). RA = LP/DT. It is normally greater than 1.85.
Bestrophins are transmembrane proteins under the control of 4 genes. The BEST1 (11q12) and BEST2 (19p13.2-p13.12) genes are expressed in the eye.
The eye diseases linked to the BEST1 gene are:
- Best's vitelliform macular dystrophy or Best's disease [MIM 153 700]:
The most common autosomal dominant macular dystrophy, but its inter- and intra-familial penetrance and expression are variable. This retinal dystrophy affects the retinal pigment epithelium and is characterized by the presence of auto-fluorescent vitelline deposits the evolutionary sequence of which is stereotyped from appearance to fragmentation of this material until its resorption. It produces a characteristic "egg yolk" image of the macula. The first signs appear in children and young adults, but can also occur later in life. The visual prognosis is usually good, in contrast to the worrying fundus images. Decline in visual acuity is usually slow and progressive; some patients present with central scotoma or metamophopsia (image distortion). Astigmatism and hyperopia are common.
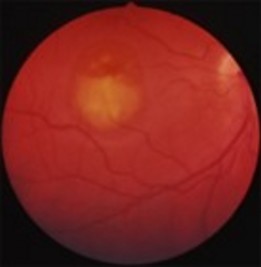
There are 6 clinical stages:
- Stage I (Previtelliform): normal vision, normal or discretely abnormal retinal pigment epithelium (central honeycomb structure) with abnormal electrooculogram.
- Stage II (Vitelliform): classic "egg yolk" lesion, with normal or slightly impaired vision.
- Stage III (Pseudohypopyon): stratification of lipofuscin. Vision similar to stage II.
- Stage IV (Vitelloruptive): the lesion is ruptured, giving a scrambled-egg appearance on the fundus. Vision similar to stages I and II, or slightly diminished.
- Stage V (Atrophic): central atrophy of the retina and retinal pigment epithelium. Significant loss of vision.
- Stage VI (Choroidal neovascularization): this complication occurs in 20 % of patients. Significant loss of vision.
Lesions are usually bilateral and relatively symmetrical, but unilateral presentations have been described. Although usually a single ocular lesion, 30 % of patients present with several lesions: this is known as multifocal Best's disease. In this case, lesions of varying sizes are found in and outside the fovea, but extrafoveal lesions are generally smaller and located superiorly in the macula. Retinal vessels cross the edges of lesions without altering their course.
- Adult-onset Vitelliform Macular Dystrophy (VMD2) [MIM 153 700] or Adult-onset Foveomacular Vitelliform Dystrophy (AFVD), which may also be caused by mutations of the PRPH2 (VMD3) genes [MIM 608 161], IMPG1 (VMD4) [MIM 616 151] and IMPG2 (VMD5) [MIM 616 152].
Cases are often sporadic. Onset between 30 and 50 years of age. No visual symptoms or slight drop in visual acuity. Fundus: autofluorescent, subfoveal vitelliform-like lesion (500-700 µm) in size.
- autosomal recessive bestrophinopathy
Usually the result of composite heterozygosity (a different mutation on each allele). Slow decline in visual acuity in the first decade. Clinical presentation: hyperopia, narrow anterior chamber with risk of increased intraocular pressure and glaucomatous optic neuropathy). Fundus: multifocal fluorescent yellow spots around the vascular arches, macular lesions with subretinal fibrosis below the fovea, macular edema and, finally, choroidal neovascularization. Pathologic ERG and electrooculogram.
- autosomal dominant vitro-retino-choroidopathy (see this term).
- MRCS syndrome (Microcornea, Rod-cone dystrophy, early-onset Cataract, posterior Staphyloma). Prevalence < 1/106. Autosomal dominant transmission. A distinction is made between MRCS 1 (mutation of the BEST1 gene) [MIM 193 220] and MRCS2 (mutation of the ALR2 gene (11q13) [MIM 619 082]. Clinical presentation: hyperopia, narrow anterior chamber, microcornea, early pulverulent cataract. The first symptoms are nyctalopia during adolescence. Visual acuity declines after 30 years of age to values between 2/10 and no light perception. Fundus: peripheral atrophy and retinal pigment epithelium abnormalities, posterior staphyloma, cone and rod dystrophy. Abnormal electrooculogram and ERG.
- some retinitis pigmentosa
Anesthetic implications:
visual disturbances, eye protection, check for associated abnormalities.
References:
-
Updated: October 2023