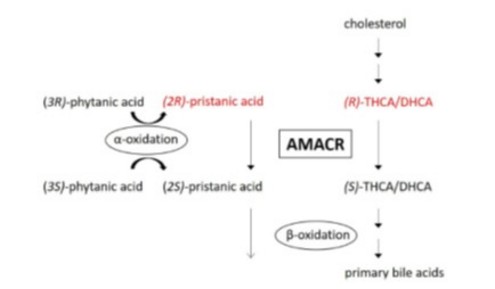
Extremely rare: fewer than 20 patients have been described. Autosomal recessive transmission of two variants of the AMACR gene (5p13.3), coding for the peroxisomal enzyme alpha-methylacyl-CoA racemase. Its deficiency leads to accumulation of toxic intermediate bile derivatives: (R)-trihydroxycholestenoic acid (THCA), (R)-dihydroxycholestenoic acid and pristanic acid (DHCA), which can cause liver and neurological problems.
It develops slowly, and is often diagnosed late, after the age of 40.
The clinical presentation is highly variable:
- retinitis pigmentosa
- peripheral sensorimotor neuropathy
- ataxia and cognitive decline
- seizures
- sometimes stroke-like episodes
- sometimes recurrent episodes of rhabdomyolysis (neuroleptic malignant syndrome?)
- rare: neonatal cholestasis, hepatic fibrosis, liver tumours
- child: asymptomatic increase in liver enzymes
- MRI: T2 hyperintense and symmetrical signal in thalamus and brainstem
Anesthetic implications:
unknown
References :
- Klouwer FCC, Roosendaal SD, Hollak CEM, Langeveld M, Poll-The BT, van Sorge AJ et al.
Redefining the phenotype of alpha-methylacyl-CoA racemase (AMACR) deficiency.
Orphanet Journal of Rare Diseases 2024Ā; 19:350 doi.org/10.1186/s13023-024-03358-9
Updated: November 2024