Très rare : < 1/106. Forme la plus sévère des maladies de surcharge en esters du cholestérol suite à un déficit en lipase acide lysosomiale. Transmission autosomique récessive d’une mutation du gène LIPA ou LAL en 10q24-q25. Ce déficit conduit à une accumulation massive de cholestérol estérifié et de triglycérides d’origine lipoprotéique dans les tissus.
Début dans les premières semaines de vie :
- distension abdominale avec hépatosplénomégalie importante, parfois ascite ; vomissements
- diarrhée par stéatorrhée
- calcifications des glandes surrénales par accumulation de cristaux de cholestérol et nécrose (signe caractéristique)
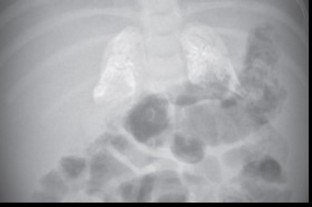
- anémie sans thrombopénie, cachexie
- décès avant 12 mois sans traitement : greffe très précoce de moelle ou de sang de cordon
Traitement substitutif: sébélipase alpha IV 1X/semaine
Récent succčs avec guérison complčte chez un nourrisson: traitement substitutif (sébélipase) vite associé au rituximab (pour éviter l’apparition d’anticorps anti-sébélipase) et suivi d’une greffe de sang de cordon.
Diagnostic : lymphocytes vacuolés, histiocytes spumeux à la ponction de moelle, très faible activité de la lipase acide (< 6%) dans les leucocytes.
Implications anesthésiques:
anémie, hépatosplénomégalie avec hypertension portale ; pas d’insuffisance corticosurrénalienne décrite malgré les calcifications surrénaliennes ; éviter une TIVA à base de propofol (surcharge en triglycérides
Références :
- Al Essa M, Nounou R, Sakati N, Le Quesne G et al.
Wolman’s disease: the King Faisal specialist hospital and research centre experience.
Annals of Saudi Medicine 1998; 18: 120-4.
- Rodriguez-Oquendo A, Kwiterovich PO.
Dyslipidaemias. In Inborn Metabolic Diseases, 5th ed, Editors : Saudubray J-M, van den Berghe G, Walter JH. Springer 2012, p 439-60.
- Ben Hassine L, Lahmar L, Louati H, Douira W, Bellagha I.
Maladie de Wolman.
Arch Pédiatr 2016 ; 23 : 1201-3.
- Krause I, Gavrielli H.
Adrenal calcifications in an infant.
NEJM 2018; 378: e36
- Eskandari SK, Revenich EGM, Pot DJ, de Boer F, Bierings M, van Spronsen FJ, van Hasselt PM, Lindemans CA, Lubout CMA.
High-dose ERT, Rituximab, and early HSCT in an infant with Wolman’s disease.
N Engl J Med 2024;390:623-9.
Mise-à-jour mars 2024