(Tyrosinose, Tyrosinémie hépatorénale)
Pathologie du métabolisme de la tyrosine (synthétisée à partir des protéines alimentaires ou tissulaires et de la phénylalanine).
Selon l’enzyme manquant on distingue :
- la tyrosinémie type I (ou tyrosinémie hépatorénale): [MIM 276 700] : transmission autosomique récessive d’une mutation du gčne FAN (15q23-q25); déficit en fumarylacétoacétate hydroxylase qui est localisée principalement dans le foie, les reins et les lymphocytes. Maladie rare (1/100.000) sauf dans certaines régions : Lac St Jean au Québec, Scandinavie. Les principales manifestations cliniques sont fort variables selon les mutations. Ce sont :
- une atteinte hépatique : hépatomégalie, forme aiguë (« crises hépatiques ») avec coagulopathie ou chronique avec cirrhose micro-et macronodulaire et risque important de carcinome hépatocellulaire
- une atteinte rénale : dysfonction tubulaire avec syndrome de Fanconi (glycosurie, aminoacidurie) ; néphromégalie avec calcifications ; glomérulosclérose ; rachitisme
- des signes neurologiques : proches de ceux d’une crise de porphyrie : crises douloureuses, opistothonos, paralysies … car une des métabolites (succinylacétone) qui s’accumule inhibe la déhydratation de l’acide δ-aminolévullinique.
-parfois une cardiomyopathie hypertrophique symétrique ou asymétrique (qui peut régresser après l’introduction du traitemernt médical) ou une hyperplasie des îlots de Langerhans (hypoglycémies).
Traitement : l’administration quotidienne de NTBC (2-2-nitro-4-trifluoromethylbenzoyl-1,3cyclohexanedione) inhibe la 4-hydroxy-phénylpyruvate diogénase et diminue rapidement la production de métabolites toxiques ; dose : 1mg/kg/jour en 2 prises et surveiller le taux plasmatique de tyrosine car s’il est > 500 µmol/L il y a un risque de toxicité oculaire (kératite, photophobie, opacités ou ulcères cornéens) ; une transplantation hépatique est réalisée avant l’âge de 2 ans (à cause du risque carcinologique) en cas de non-réponse.
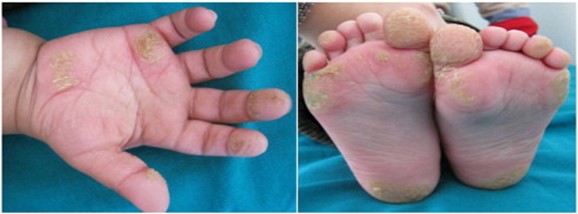
- la tyrosinémie type II (ou tyrosinémie oculocutanée, ou syndrome de Richner-Hanhart) : [MIM 276 600]: transmission autosomique récessive d’une mutation du gčne TAT (16q21.1) qui entraîne un déficit en tyrosine aminotransférase ; trčs rare, observé principalement dans les populations arabes et méditerranéennes ; signes oculaires (75%) : photophobie, larmoiement, ulcčres cornéens ; signes cutanés (80%): hyperkératose palmoplantaire douloureuse ; retard de croissance ; parfois retard mental léger (60%)
Traitement : régime pauvre en protéines et en phénylalanine.
- la tyrosinémie type III (ou tyrosinose) : [MIM 276 710]: transmission autosomique récessive d’une mutation du gčne HPD (12q14-qter) ; déficit en 4-hydroxyphenylpyruvate dioxygénase ; manifestations cliniques variables : aucun signe clinique ou ataxie, léthargie, retard mental. Traitement : régime pauvre en protéines et en phénylalanine.
- la tyrosinémie transitoire du nouveau-né : immaturité transitoire de la 4-hydroxy-phénylpyruvate diogénase (surtout chez le prématuré ne recevant pas de suppléments de vitamine C)
Implications anesthésiques:
- type I : vérifier les fonctions hépatique et rénale ; monitorer la glycémie ; sauf si le traitement au NTBC est efficace, éviter les agents qui peuvent déclencher une crise de porphyrie aiguë et le N2O. Transplantation hépatique ou hépato-rénale .
- type II : éviter les lumières vives, protection oculaire ; éviter un apport excessif de protéines exogènes (aspirer le sang dégluti)
Références :
- Russo PA, Mitchell GA, Tanguay RM.
Tyrosinemia: a review.
Pediatr Developmental Pathol 2001; 4: 212-21.
- Madan N, Arnon R, Arnon R.
Evaluation of cardiac manifestations in pediatric liver transplantation candidates.
Pediatr Transplantation 2012; 16: 318-28.
Mise-à-jour juin 2022