Groupe des pathologies musculaires (muscle squelettique et ou cardiaque) causées par une mutation du gčne de la titine (TTN (2q31.2). La titine ou connectine est la plus grande protéine du corps humain, et est la protéine la plus abondante dans le muscle strié aprčs la myosine et l’actine. Cette protéine élastique liée aux filaments de myosine est le principal constituant des filaments longitudinaux qui assurent le maintien de l’architecture myofibrillaire. Au niveau de la bande A, la titine possčde de multiples sites de fixation avec la myosine, la protéine C ou l’AMP désaminase. Au niveau de la bande I, elle est liée ŕ la calpaďne 3. Au niveau de la strie Z, son association avec l’actine garantit le maintien de l’assemblage myofibrillaire au cours de la contraction musculaire.
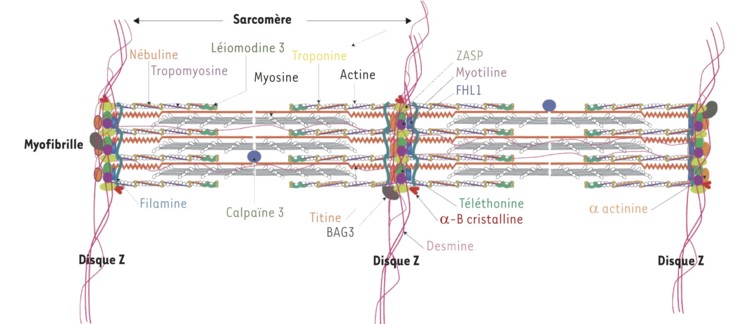
Plus de 127 mutations du gčne TNT, associées ŕ au moins 10 phénotypes différents (myopathie, cardiomyopathie ou les 2), selon qu’un isoforme musculaire squelettique et ou cardiaque est atteint, étaient décrites en 2020.Ces mutations peuvent entraîner des cardiomyopathies dilatées ou hypertrophiques, des dysplasies ventriculaires droites arythmogčnes, une dizaine de phénotypes différents de myopathie.
Atteintes cardiaques : on distingue,
- cardiomyopathie hypertrophique sarcomérique (<5%), transmission autosomique dominante : hypertrophie asymétrique du VG (en général au niveau du septum interventriculaire), risque de mort subite (environ 1%/an)
- cardiomyopathie dilatée (CMD1G)(15-25%), transmission autosomique dominante : dilatation du VG ou biventriculaire associée ŕ une dysfonction systolique ; évolution vers l’insuffisance cardiaque
- cardiomyopathie restrictive
- cardiomyopathie ventriculaire arythmogčne (voir ce terme)
- plus rarement : non-compaction du ventricule gauche (voir ce terme), CIA, CIV
Atteintes musculaires (tableau): on distingue,
A. Les formes ŕ début congénital ou infantile : souvent début anténatal (immobilité fśtale, arthrogrypose). En général, retard moteur et faiblesse musculaire non sélective, avec une atteinte relativement diffuse, ŕ prédominance proximale et axiale (muscles paravertébraux, diaphragme), et une composante rétractile (rétractions des tendons d’Achille, Rigid Spine). L’atteinte myocardique primaire (cardiomyopathie dilatée) n’est pas toujours présente.
- myopathie multi-minicore (voir ce terme) autosomique récessive avec atteinte cardiaque (MmD with heart disease) qui inclut l’EOMFC (Early-onset Myopathy with Fatal Cardiomyopathy): image de myopathie congénitale avec cores, cardiomyopathie dilatée progressive, décčs dans l’adolescence et un phénotype de myopathie d’Emery-Dreifuss
- dystrophie des ceintures LGMDJ2 ou LGMDR10
- arthrogryposis multiplex congenita (voir ce trerme) et non-compaction du ventricule gauche.
B. les formes qui débutent aprčs 10 ans (juvénile) ou ŕ l’âge adulte : histologie ; dystrophie ou images myofibrillaires
- dystrophie musculaire tibiale de type Udd (Finlande) (TMD Tibial Muscular Dystrophy)[MIM 188 840] : steppage
- HMERF (Hereditary Myopathy with Early Respiratory Failure ou myopathie d’Edström): apparition d’un syndrome restrictif ŕ l’adolescence, tétraparésie. CPK nl
- Myopathie Emery-Dreifuss-like: CPK élevés (10x), contractures, pas d’atteinte cardiaque
- titinopathie de type Emery-Dreifuss (voir ce terme) autosomique récessive
- titinopathie distale juvénile autosomique récessive
- myopathies myofibrillaire
- myopathie en collier (necklace)
A l’histologie, on peut observer, selon le phénotype : lésions dystrophiques, vacuoles bordées, cores, centralisation nucléaire, amas myofibrillaires, inclusions éosinophiles (en lacet), une prédominance des fibres de type 1, une irrégularité de la taille des fibres.
|
nom |
transmission |
âge de début |
phénotype |
biopsie
|
|
Dystrophie musculaire tibiale |
AD |
>35 ans |
déficit des releveurs des pieds |
fibrose modérée vacuoles bordées |
|
Dystrophie musculaire tibiale ŕ début précoce |
AR |
20 ans |
déficit des releveurs des pieds |
dystrophie |
|
Myopathie des ceintures LGMDJ2 ou LGMDR10 |
AR |
<20 ans |
déficit trčs évolutif des ceintures pelviennes et scapulaires |
vacuoles bordées |
|
Myopathie héréditaire avec atteinte respiratoire précoce (HMERF) |
AD (rares AR) |
>20 ans |
atteinte distale puis proximo-distale Trčs évolutive Atteinte respiratoire précoce |
Corps cytoplasmiques Vacuoles bordées |
|
Myopathie Emery Dreifuss-like |
AR |
<10 ans |
déficit des ceintures Rétractions d’apparition précoce « rigid Spine » atteinte respiratoire |
Corps cytoplasmiques Vacuoles bordées Absence de minicores |
|
Myopathie précoce avec cardiopathie létale (EOMFC) |
AR |
néonatale |
déficit moteur modéré ptosis, parésie faciale pseudo-hypertrophie des mollets cardiomyopathie dilatée sévčre et précoce évolution rapide. décčs < 20 ans |
Aspect dystrophique Minicores |
|
Myopathie congénitale ŕ multiminicores avec cardiomyopathie |
AR |
<20 ans |
déficit ŕ prédominance axiale Rétractions diffuses avec « rigid spine |
Minicores Dépôts basophiles en forme d’étoile |
|
Myopathie 'centronucléaire' |
AR |
<20 ans |
déficit axial et proximal modéré Parésie faciale Syndrome restrictif Absence de cardiomyopathie |
Noyaux centraux dans la plupart des fibres musculaires Minicores |
|
Dystrophie musculaire proximale |
AR |
adulte |
déficit proximal moins sévčre que LGMD2J |
Aspect dystrophique |
|
LGMD avec cardiomyopathie |
AD |
adulte |
Cardiomyopathie sévčre et LGMD modéré |
Minicores |
|
Myopathie distale des membres inférieurs ŕ prédominance postérieure |
AD |
adulte |
déficit distal avec évolution proximale Atteinte des mollets |
Noyaux centraux Minicores |
|
arthrogrypose |
AR |
prénatal |
Contractures, avec ou sans cardiomyopathie |
|
Implications anesthésiques:
échocardiographie ; vérification des CPK ; traitement de l’insuffisance cardiaque ; parfois défibrillateur automatique implanté. En cas d’atteinte musculaire, considérer comme ŕ risque de rhabdomyolyse induite par les halogénés et la succinylcholine.
Références :
- Juntas-Morales R, Perrin A, Cossée M.
Corrélation phénotype-génotype dans les titinopathies.
Cahiers de Myologie 2020; 21: 16-20.
- Oates EC, Jones KJ, Donkervoort S, Charlton A et al.
Congenital titinopathy : comprehensive characterization and pathogenic insights.
Ann Neurol 2018 ; 83 : 1105-24
- Wacker J, Di Bernardo S, Lobrinus JA, et al.
Successful heart transplant in a child with congenital core myopathy and delayed-onset restrictive cardiomyopathy due to recessive mutations in the titin (TTN) gene.
Pediatric Transplantation 2023; e14561. doi:10.1111/petr.14561
Mise-à-jour mars 2024