Voir aussi Phénylcétonurie
- Formes avec hyperphénylalaninémie ou Phénylcétonurie type 2
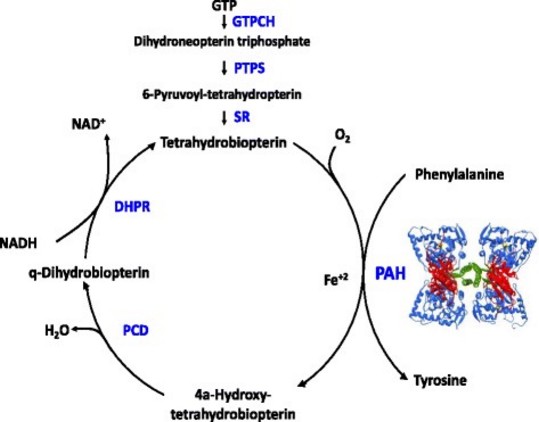
Transmission autosomique récessive d‘une mutation d’un des gčnes qui assurent la synthčse ou la régénération de la tétrahydrobioptérine (BH4), un cofacteur de la phénylalanine hydroxylase, de la tyrosine hydroxylase, de la tryptophane hydroxylase (2 isoformes), de l’alkylglycérol mono-oxygénase, et de la NO synthase (3 isoformes): ce sont GCH1 (GTP cyclohydroxylase I en 14q22.2) [MIM 233 910], PTS (6- pyruvoyl-tétrahydroptérine synthase en 11q23.1) (54%)[MIM 261 640], PCBD (ptérine-4á-carbinolamine déhydratase en 10q22.2 [MIM 264 070], ou QDPR (dihydroptéridine réductase en 4p15.32) (33%) [MIM 261 630].
Le déficit en BH4 dű ŕ une mutation d’un de ces gčnes provoque une forme de phénylcétonurie (1-2% des cas), car outre l’hyperphénylalanémie le dysfonctionnement de toutes ces enzymes entraîne un déficit en neurotransmetteurs monoaminergiques : dopamine, sérotonine et NO.
La phénylcétonurie fait l'objet d'un dépistage systématique quelques jours aprčs la naissance (test de Guthrie). En cas de taux pathologique de Phe, un test de réponse ŕ l’administration orale de BH4 est réalisé: 20 mg/kg/j pendant 48h. Si on observe une diminution des taux sanguins de Phe de > 30% du taux initial, un traitement ŕ base de BH4 et d’un régime (souvent moins sévčre qu’en cas de phénylcétonurie classique type 1) proposé.
Sans traitement, cette forme d’hyperphénylalaninémie entraîne :
- une hypotonie et un retard mental sévčre
- une dystonie, un tremblement au repos, des mouvements désordonnés, une rigidité extrapyramidale avec bradykinésie
- des troubles du comportement : hyperactivité, troubles de l’attention, anxiété, troubles obsessionnels compulsifs, psychose,
- des troubles du sommeil, une salivation excessive
- parfois : spasmes en flexion, épilepsie
- des épisodes d’hyperthermie.
Il y a également une hypomélanose (peau claire, cheveux blonds, yeux bleus, eczéma) et une odeur caractéristique dite « de souris ».
En cas de déficit en ptérine-4á-carbinolamine déhydratase, on observe trčs souvent une hypomagnésémie et un diabčte précoce de type MODY3 (voir ce terme).
En cas d’hyperphénylalaninémie sans mutation de la phénylalanine hydroxylase ni anomalie du métabolisme de la tétrahydroptérine, il faut rechercher une mutation du gčne de la DNAJC12, une protéine chaperone qui contribue au déploiement de la phénylalanine hydroxylase.
Trois présentations cliniques :
- asymptomatique : hyperphénylalaninémie découverte lors du test de Guthrie, ce qui permet le diagnostic et un traitement précoce
- symptomatique (dégradation neurologique) malgré un régime bien suivi : on a cru ŕ tort ŕ une hyperphénylalaninémie isolée (type 1)
- symptomatique sans régime : faux négatif ŕ la naissance
Outre un régime qui consiste en une alimentation restreinte en Phe associée ŕ la prise de substituts d’acides aminés, cette pathologie nécessite l’administration de :
- tétrahydrobioptérine (BH4), cofacteur de la phénylalanine-hydroxylase, ce qui autorise un régime alimentaire plus léger. La dose (saproptérine dihydrochloride ou Kuvan®) est de 5 ŕ 20 mg/kg/j en 2-3 prises. On évite cependant les suppléments de BH4 en cas de déficit en dihydroptéridine réductase car il y a un risque d’excčs de bihydrobioptérine.
- acide folique : 15 mg/j
- 5-hydroxytryptophane : de 1 ŕ 10 mg/kg/j en 4 prises, sauf en cas de déficit en ptérine-4á-carbinolamine déhydratase ou de déficit en guanosine triphosphate cyclohydrolase
- L-Dopa associée ŕ un inhibiteur de la dopa-décarboxylase : 1-2 mg/kg/j en 4 prises
et, dans certains cas :
- entacapone (inhibiteur de la catéchol-o-méthyl-transférase): 15 µg/kg/j en 2-3 prises
- sélégiline (inhibiteur de l’IMAO B) : de 0,1 ŕ 0,25 mg/kg/j en 3-4 prises (ne pas co-administrer avec le 5-hydroxytryptophane : risque de syndrome sérotoninetgique)
- pramipexole (agoniste des récepteurs ŕ la dopamine): de 6 ŕ 35 µg/kg/j en 2 prises.
Attention : l’aspartame et la gélatine sont ŕ éviter chez les enfants atteints de
phénylcétonurie. L’aspartame est l’ester méthylique de l’aspartate Phe qui se scinde dans l’organisme en deux acides aminés, l’acide aspartique et la Phe, et en méthanol. La gélatine, qui entre souvent dans la composition des gélules, est particuličrement riche en protéines (86 g/100 g de produit). Ces deux substances sont trčs souvent des composants des médicaments. Il faut donc veiller ŕ éviter les médicaments contenant ces substances. La quantité apportée de Phe par ces deux substances reste cependant faible. Le médicament peut donc ętre administré en surveillant le taux et en diminuant la quantité de Phe dans l’alimentation (contact nécessaire avec la diététicienne) si un traitement est vraiment nécessaire et qu’il n’existe pas d’alternative sans aspartame ou sans gélatine. Il est toujours utile de vérifier dans un compendium la composition d’un médicament (liste des excipients).
2) Formes sans hyperphénylalaninémie
Deux formes:
- la transmission autosomique dominante d’une mutation du gčne GCH1 (14q22.2) qui entraîne un déficit en guanosine triphosphate cyclohydrolase I [MIM 128 230]: c’est la cause la plus fréquente de la dystonie répondant ŕ la dopa, un syndrome caractérisé par une dystonie fluctuante (qui s’aggrave au cours de la journée) aussi appelé syndrome de Segawa autosomique dominant (DYT5a) (voir ce terme)
- la transmission autosomique récessive d’une mutation du gčne SPR (2p13.2) qui entraîne un déficit en sepiaptérine réductase [MIM 612 716]
Implications anesthésiques:
Dans les formes avec hyperphénylalaninémie, il faut appliquer les précautions nécessaires pour éviter l’augmentation des taux sanguins de Phe, comme :
- contacter l’endocrinologue pour avis et connaître le taux habituel de Phe chez le patient
- chez l’adulte, vérifier les fonctions cardiaque et rénale
- toute infection augmente le taux sanguin de phénylalanine et nécessite une adaptation des apports en Phe
- éviter le catabolisme protéique : durée du jeűne préopératoire courte et assurer un apport suffisant de glucose en périopératoire ; monitorage de la glycémie.
- du fait du risque de déficience en vitamine B12 si le régime alimentaire n'est pas adéquatement supplémenté, il est prudent d'éviter le N2O.
- en cas d’alimentation parentérale : utiliser une solution sans phénylalanine.
- colloďdes ŕ base de gélatines : bien que leur élimination se fasse principalement sous forme urinaire inchangée, il faut les utiliser avec prudence.
- en cas de risque d’ingestion de sang (chirurgie oropharyngée : amygdalectomie, végétations, fente palatine, stomatologie) il est utile de placer un packing pharyngé et de vider l’estomac avant le réveil car le sang dégluti est riche en protéines.
Dans toutes les formes, il faut tenir compte des interactions médicamenteuses entre les agents anesthésiques et le traitement chronique du patient:
- L-dopa: la prise chronique de L-dopa pourrait entraîner une hypotension orthostatique et diminuer la réponse aux vasopresseurs indirects comme l’éphédrine; il est donc plus prudent d’utiliser des doses titres de vasopresseurs ŕ effet direct. Il faut également éviter les antagonistes centraux de la dopamine comme le métoclopramide et les butyrophénones.
- pramipexole (agoniste des récepteurs dopaminergiques)
- 5-hydroxytryptophane
- entacapone (un inhibiteur de la COMT) qui augmente la sensibilité de la réponse aux vasopresseurs directs
- sélégiline (inhibiteur de type B de la MAO) in 3-4 doses: en principe pas d’interaction avec les agents anesthésiques ŕ moins de 10 mg/j mais se comporterait comme une IMAO A ŕ doses plus fortes.
On ignore si les sétrons (inhibiteurs 5HT3) peuvent ętre utilises comme antiémétiques. Il est preferable d’éviter le tramadol ŕ cause de son mode d’action monoaminergique. Eviter le trimétroprim/sulfamétroxazole.
De plus, il y a un risque de troubles du comportement et d’hyperthermie en post-opératoire.
Références :
- Dal D, Celiker V.
Anaesthetic management of a strabismus patient with phenylketonuria.
Paediatr Anaesth 2003; 13: 740-1. et correspondance: Pediatr Anesth 2004; 14: 701-2.
- Feillet F, Bonnemains C.
La phénylcétonurie : nouveaux traitements.
Arch Pédiatr 2013 ; 20 : 1165-8.
- Wyatt SS, Gill RS.
An absolute contraindication to nitrous oxide.
Anaesthesia 1999 ; 54 : 307.
- Walter JH, Lachmann RH, Burgard P.
Hyperphenylalaninaemia,
In Inborn Metabolic Diseases, 5th edition, Saudubray, van den Berghe, Walter, Springer 2011, p 251-64.
- Opladen T, Lopez-Laso E, Cortes-Saladelafont E, Pearson TS, Sivri HS et al.
Consensus guidelines for the diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies.
Orphanet J Rare Diseases 2020; 15:126
Mise à jour: août
2020