[MIM 105 400, 205 250, 300 857, 606 070, 606 640, 608 030, 608 031, 608 627]
(maladie de Charcot, maladie de Lou Gehrig)
Rare: prévalence 1/20.000 en Europe de l’Ouest, plus élevée dans des îles du Pacifique Ouest. Affection neurodégénérative due ŕ une dégénérescence des motoneurones du cortex moteur, de la voie corticospinale, du tronc cérébral et de la moelle épiničre. Début habituel vers 60 ans. Cas sporadiques dans la majorité des cas, mais 5 ŕ 10% des cas sont familiaux.
On distingue :
- la forme classique, sporadique : différents facteurs favorisants ont été identifiés comme le tabagisme, les microtraumatismes cérébraux répétés (sports de contact, boxeurs)
- les formes familiales :
- type 1 : transmission autosomique dominante ou récessive d’une mutation du gčne SOD1 (21q22.11) qui code la superoxide dismutase Cu/Zn [MIM 105 400]
- type 2 ou juvénile : transmission autosomique récessive d’une mutation du gčne ALS2 (2q33.1) qui code l’alsine [MIM 105 100]
- type 3 : transmission autosomique dominante d’une mutation d’un gčne en 18q21 [MIM 606 640]
- type 4 ou juvénile: transmission autosomique dominante d’une mutation du gčne SEXT1 (9q34.14) qui code la sénataxine [MIM 602 433]
- type 5 ou juvénile : transmission autosomique récessive d’une mutation du gčne SPG11 (15q21.1) qui code la spatacsine [MIM 602 099]
- type 6, avec ou sans démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne FUS (16p11.2) [MIM 608 030]
- type 7 : transmission autosomique dominante d’une mutation d’un gčne situé en 20p13 [MIM 608 031]
- type 8 : transmission autosomique dominante d’une mutation du gčne VAPB (20q13.32) qui code une protéine associée ŕ la membrane des vésicules [MIM 608 627]
- type 9 : transmission autosomique dominante d’une mutation du gčne ANG (14q11.2) qui code l’angiogénine [MIM 611 895]
- type 10, avec ou sans démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne TARDBP (1p36.22) [MIM 612 069]
- type 11 : transmission autosomique dominante d’une mutation du gčne FIG4 (6q21) [MIM 612 577]
- type 12 : transmission autosomique dominante ou récessive d’une mutation du gčne OPTN (10p13) qui code l’optineurine [MIM 613 435]
- type 13 : transmission autosomique dominante d’une mutation du gčne ATXN2 (12q24.12) qui code l’ataxine 2 [MIM 183 090]
- type 14, avec ou sans démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne VCP (9p13.3) qui code une protéine associée ŕ la valosine [MIM 613 954]
- type 15, avec ou sans démence fronto-temporale : transmission liée ŕ l’X d’une mutation du gčne UBQLN2 (Xp11.21) qui code l’ubiquiline 2 [MIM 300 857]
- type 16 ou juvénile : transmission autosomique récessive d’une mutation du gčne SIGMAR1 (9p13.3) [MIM 614 373]
- type 17 : transmission autosomique dominante d’une mutation du gčne CHMP2B (3p11.2) [MIM 614 696]
- type 18 : transmission autosomique dominante d’une mutation du gčne PFN1 (17p13.2) qui code la profiline 1[MIM 614 808]
- type 19 : transmission autosomique dominante d’une mutation du gčne ERBB4 (2q34) MIM 615 515]
- type 20 : transmission autosomique dominante d’une mutation du gčne HNRNPA1 (12q13.13) qui code une protéine ribonucléaire du noyau [MIM 615 426]
- type 21 : transmission autosomique dominante d’une mutation du gčne MATR3 (5q31.2) qui code la matrine 3 [MIM 606 070]
- type 22, avec ou sans démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne TUBA4A (2q35) qui code la tubuline alpha 4A [MIM 606 070]
- type 23 : transmission autosomique dominante d’une mutation du gčne ANXA11 (10q22.3) qui code l’annexine A11[MIM 617 938]
- type 24 : susceptibilité ŕ la sclérose latérale amyotrophique suite ŕ une mutation du gčne NEK1 (4q33) [MIM 617 892]
- type 25 : susceptibilité ŕ la sclérose latérale amyotrophique suite ŕ la transmission autosomique dominante d’une mutation du gčne KIF5A (12q13.3) [MIM 617 921]
- forme associée ŕ NEFH : transmission autosomique dominante ou récessive d’une mutation du gčne NEFH (22q12.2) qui code un neurofilament [MIM 105 400]
- forme associée ŕ PRPH : transmission autosomique dominante d’une mutation du gčne PRPH (12q13.12) qui code la périphérine [MIM 105 400]
- forme associée ŕ la dynactine 1 : transmission autosomique dominante d’une mutation du gčne DCTN1 (2p13.1) qui code la dynactine 1 [MIM 105 400]
- sclérose latérale amyotrophique et/ou démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne C9orf72 (9p21.2) [MIM 105 550]
- sclérose latérale amyotrophique et/ou démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne CHCHD10 (22q11.23) [MIM 615 911]
- sclérose latérale amyotrophique et/ou démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne SQSTM1 (5q35.3) [MIM 664 37]
- sclérose latérale amyotrophique et/ou démence fronto-temporale : transmission autosomique dominante d’une mutation du gčne TBK1 (12q14.2) [MIM 616 439].
- la forme dite du Pacifique-Ouest : dans l’île de Guam, elle semble liée ŕ l’accumulation de béta-méthylamino-L-alanine dans le tissu cérébral ; cette molécule est un neurotoxique produit par une algue bleue-verte qui réside sur les racines et dans les semences du sagoutier (cycad).
Physiopathologie mal connue : destruction progressive des motoneurones suite à la diminution de l’élimination du glutamate par les astrocytes : aggrégation de protéines dans le cytoplasme ou les axones ; perte du contrôle inhibiteur GABAergique des motoneurones corticaux etc…
La symptomatologie est très variable et dépend de la topographie des neurones moteurs atteints ; l’atteinte peut être initialement unilatérale ou localisée :
* motoneurones spinaux (2/3 des cas) : faiblesse musculaire puis amyotrophie ; spasticité ; fasciculations ; hyperréflexie ; clonus
* motoneurones bulbaires : dysarthrie, dysphagie, spasmes laryngés, sialorrhée
* en cas d’atteinte bilatérale des faisceaux cortico-bulbaires : crises émotionnelles : rires et pleurs paradoxaux
* l’atteinte des muscles respiratoires entraîne une respiration paradoxale, une tachypnée avec soif d’air ; des apnées obstructives du sommeil
* il n’y a pas de troubles sensitifs ni du contrôle des sphincters ; les fonctions cognitives sont conservées
Diagnostic : EMG
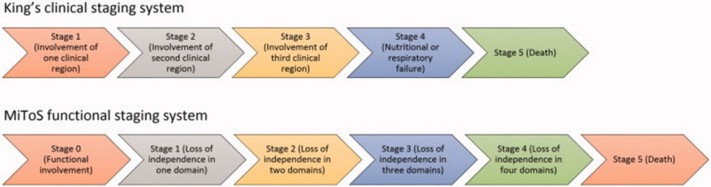
L’évolution de la maladie est évaluée en utilisant la classification du King’s College ou l’échelle MiToS:
Pronostic : sombre : insuffisance respiratoire progressive avec décès dans les 3 à 5 ans.
Traitement :
- symptomatique : kiné, VNI, gastrostomie…
- antispastiques : baclofène, dantrolène, tizanidine, toxines botuliques
- riluzole : qui diminue la taux de glutamates au niveau des terminaisons nerveuses, notamment en bloquant les récepteurs NMDA post-synaptiques ; parfois augmentation des enzymes hépatiques, HTA
- hautes doses vitamine B12
- la ceftriaxone, une béta-lactamine qui augmente un des transporteurs du glutamate
- dexpramipexole : un antioxydant utilisé comme anti-Parkinsonien
- pacing du diaphragme ?
Implications anesthésiques:
évaluer la fonction respiratoire : SpO2 ŕ l’air, épreuves fonctionnelles, toux ; risque de fausse déglutition : extubation ŕ l’éveil complet; éviter la succinylcholine : des cas d’hyperkaliémie ont été décrits. En cas de VNI, il faut la poursuivre en post-opératoire immédiat ; en cas d’insuffisance respiratoire majeure, utilisation prudente d’un supplément d’O2. Curarisation avec prudence: un cas d’antagonisation incomplčte du rocuronium par le sugammadex a été décrit.
Plusieurs cas d‘anesthésie péridurale lombaire ou de rachianesthésie combinés ŕ une sédation (propofol, dexmédétomidine ou rémimazolam) ont été décrits sans aggravation de l’évolution clinique.
Références :
- Kwon Y-S, Lim Y-H, Woo S-H, Yon JH, Kim K-M. Epidural anesthetic management of a patient with amyotrophic lateral sclerosis (ALS). Korean J Anesthesiol 2009; 57: 361-3
- Prabhakar A, Owen CP, Kaye AD.
Anesthetic management of the patient with amyotrophic lateral sclerosis.
J Anesth 2013; 27: 909-18.
- Sertöz N, Karaman S.
Peripheral nerve block in a patient with amyotrophic lateral sclerosis.
J Anesth 2012; 26: 314-5.
- Turner M, Lawrence H, Arnold I, Ansorge O, Talbot K.
Catastrophic hyperkalaemia following administration of suxamathonium chloride to a patient with undiagnosed amyotrophic lateral sclerosis.
Clin Med 2011; 11: 292-3
- Thourot M, Cornillon B, Robert R, Royer D, Patrigeon R-G.
Hyperkaliémie menaçante suite ŕ une injection de succinylcholine orientant le diagnostic vers une sclérose latérale amyotrophique.
Anesth Réanim 2017 ; 3 : 193-7.
- Fang T, Al Khleifat A, Stahl DR, Lazo la Torre C, Murphy C et al.
Comparison of the King’s and MiToS staging systems for ALS.
Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration 2017; 18: 227-32
- Benarroch L, Bonne G, Rivier F, Hamroun D.
The 2020 version of the gene table of neuromuscular disorders. Neuromusc Dis 2019 ; 29 : 980-1018 ou http://www.musclegenetable.fr .
- Hoeper AM, Barbara DW, Watson JC, Sprung J, Weingarten T.
Amyotrophic lateral sclerosis and anesthesia: a case series and review of the literature.
J Anesth 2019; 33: 257-65.
- Panchamia JK, Gurrieri C, Amundson AW.
Spinal anesthesia for amyotrophic lateral sclerosis patient undergoing lower extremity orthopedic surgery: an overview of the anesthetic considerations.
International Medical Case Reports Journal 2020 ; 13 : 249-54
- Chun HR, Chung J, Kim NS, Kim AJ, Kim S, Kang KS.
Incomplete recovery from rocuronium-induced muscle relaxation in patients with amyotrophic lateral sclerosis using sugammadex: A case report.
Medicine 2020;99:3 (e18867).
- Nimma S, Gans A, MD,Wardhan R, Allen W.
Remimazolam sedation and neuraxial anesthesia in a patient with amyotrophic lateral sclerosis undergoing an open colectomy: a case report.
A&A Practice 2023;17:e01733
Mise-à-jour janvier 2024