[MIM 261 600]
(Oligophrénie phénylpyruvique, Phénylcétonurie type 1, maladie de Folling)
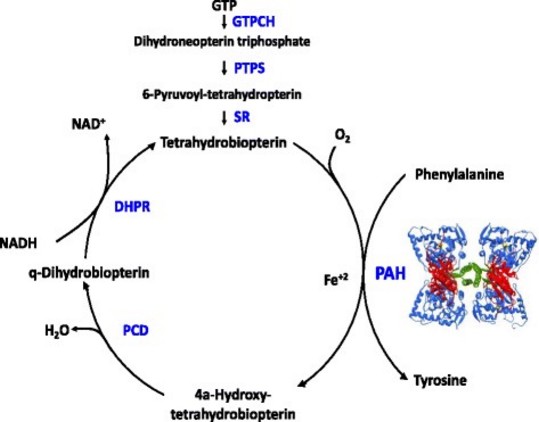
Prévalence : 1/18.000, mais varie de 1/2600 en Turquie ŕ 1/50.0000 en Grande Bretagne et 1/100.000 au Japon. Transmission autosomique récessive : 98% des cas sont dus ŕ une mutation (plus de 950 variants décrits) du gčne PAH (phénylalanine hydroxylase qui intervient dans la synthčse de T-Tyrosine et ainsi de la L-dopa et de noradrénaline) en 12q23.2. Une minorité de cas (phénylcétonurie type 2) est due ŕ une mutation d’un des gčnes qui assure la synthčse ou la régénération de la tétrahydrobioptérine (BH4) : GTPCH, PTPS, PCD, DHPR (voir Déficit en tétrahydrobioptérine).
La phénylcétonurie fait l'objet d'un dépistage systématique quelques jours aprčs la naissance (test de Guthrie). Le traitement ŕ vie, qui consiste en une alimentation restreinte en Phe associée ŕ la prise de substituts d’acides aminés, est instauré si le taux sanguin de Phe dépasse 360 µmol/l. Taux cible : 120 ŕ 360 µmol/L. Taux cible : 120 ŕ 360 µmol/L avant l’âge de 12 ans ou en cas de grossesse, 120 ŕ 600 µmol/L aprčs l’âge de 12 ans.
Les autres causes d’hyperphénylalaninémie doivent ętre exclues (prématurité, insuffisance hépatique, anomalie du métabolisme de BH4). Un test de réponse ŕ l’administration orale de BH4 est réalisé: 20 mg/kg/j pendant 48h. Si on observe une diminution des taux sanguins de Phe de > 30% du taux initial, un traitement ŕ base de BH4 et d’un régime moins sévčre est proposé (voir Déficit en tétrahydrobioptérine). En cas d’hyperphénylalaninémie sans mutation de la phénylalanine hydroxylase ni anomalie du métabolisme de la tétrahydroptérine, il faut rechercher une mutation du gčne de la DNAJC12, une protéine chaperone qui contribue au déploiement de la phénylalanine hydroxylase.
Sans régime alimentaire, l'affection entraîne un retard mental sévčre (troubles du comportement, psychoses, spasmes en flexion, épilepsie) et une hypomélanose (peau claire, cheveux blonds, yeux bleus) ainsi qu’une odeur caractéristique dite « de souris ».
Il faut donc y penser devant ces manifestations chez les enfants provenant de pays oů le dépistage néonatal n’est pas pratiqué.
Le phénotype est trčs variable et dépend de l’activité enzymatique résiduelle et de la sensibilité ŕ la prise de BH4.
On distingue 4 formes d’hyperphénylalaninémie en fonction de la tolérance ŕ la Phe, c’est-ŕ-dire la dose quotidienne maximale de Phe d’origine alimentaire qui permet de garder les taux sanguins de Phe entre 120 ŕ 360 µmol/L :
- classique ou sévčre: 250-350 mg de Phe/jour
- modérée:350-400 mg de Phe/jour
- bénigne 400-600 mg de Phe/jour
Un nouveau traitement permet de traiter ces patients sans régime alimentaire. Il s’agit de la pegvaliase (Palynziq® BioMarin, Novato, CA), une forme injectable de phénylalanine ammonia lyase, une enzyme qui métabolise la phénylalanine sanguine et remplace l’activité déficitaire de la phénylalanine hydroxylase. Dose par voie sous-cutanée: 2,5 ŕ 40 mg/semaine. Objectif thérapeutique : taux de Phe sanguin entre 30 et 360 µmol/L.
On parle d’hyperphénylalaninémie non-phénylcétonurique quand le taux sanguin de Phe reste < 600 µmol/L avec un régime alimentaire normal. Ces patients ont un développement et un comportement normaux et la nécessité de leur imposer un régime est un sujet de controverse.
Attention : l’aspartame et la gélatine sont à éviter chez les enfants atteints de phénylcétonurie. L’aspartame est l’ester méthylique de l’aspartate de phénylalanine qui se scinde dans l’organisme en deux acides aminés, l’acide aspartique et la phénylalanine, et en méthanol. La gélatine, qui entre souvent dans la composition des gélules, est particuličrement riche en protéines (86 g/100 g de produit). Ces deux substances sont trčs souvent des composants des médicaments. Il faut donc veiller ŕ éviter les médicaments contenant ces substances. La quantité apportée de phénylalanine par ces deux substances reste cependant faible. Le médicament peut donc ętre administré en surveillant le taux et en diminuant la quantité de phénylalanine dans l’alimentation (contact nécessaire avec la diététicienne) si un traitement est vraiment nécessaire et qu’il n’existe pas d’alternative sans aspartame ou sans gélatine. Il est toujours utile de vérifier dans un compendium la composition d’un médicament (liste des excipients).
Chez l’adulte qui suit un régime pauvre en Phe depuis la naissance on observe:
- une dyslipidémie athérogčne, une tendance au surpoids et une augmentation de la rigidité des parois artérielles
- une protéinurie et une diminution de la filtration glomérulaire
Implications anesthésiques:
- contacter l’endocrinologue pour avis et connaître le taux habituel de Phe chez le patient ; les implications d’un traitement par la pegvaliase ne sont pas encore connues
- chez l’adulte, vérifier les fonctions cardiaque et rénale
- toute infection augmente le taux sanguin de phénylalanine et nécessite une adaptation des apports en Phe
- éviter le catabolisme protéique : durée du jeűne préopératoire courte et assurer un apport suffisant de glucose en périopératoire; monitorage de la glycémie.
- du fait du risque de déficience en vitamine B12 si le régime alimentaire n'est pas adéquatement supplémenté, il est prudent d'éviter le N2O.
- en cas d’alimentation parentérale : utiliser une solution sans phénylalanine.
- colloďdes ŕ base de gélatines : bien que leur élimination se fasse principalement sous forme urinaire inchangée, il faut les utiliser avec prudence.
- en cas de risque d’ingestion de sang (chirurgie oropharyngée : amygdalectomie, végétations, fente palatine, stomatologie) il est utile de placer un packing pharyngé et de vider l’estomac avant le réveil car le sang dégluti est riche en protéines.
Références :
- Dal D, Celiker V.
Anaesthetic management of a strabismus patient with phenylketonuria.
Paediatr Anaesth 2003; 13: 740-1. et correspondance: Pediatr Anesth 2004; 14: 701-2.
- Feillet F, Bonnemains C.
La phénylcétonurie : nouveaux traitements.
Arch Pédiatr 2013 ; 20 : 1165-8.
- Van Wegberg AMJ, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, Bosch AM, Burlina A et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet Journal of Rare Diseases 2017; 12:62
- Mahan KC, Gandhi MA, Anand S.
Pegvaliase: a novel treatment option for adults with phenylketonuria.
Curr Med Res Opin 2019;35:647-51. doi: 10.1080/03007995.2018.1528215.
- Cunningham A, Rohr F, Splett P, Mofidi S, Bausell H, Stembridge A, Kenneson A, Singh RH.
Nutrition management of PKU with pegvaliase therapy: update of the web‑based PKU nutrition management guideline recommendations.
Orphanet Journal of Rare Diseases (2023) 18:155 doi.org/10.1186/s13023-023-02751-0
Mise à jour juillet 2023