(syndrome de Peters)
Transmission autosomique récessive. Fait partie d'un groupe d'anomalies oculaires qui sont la conséquence d'un trouble du développement embryonnaire de l'oeil. Elles sont regroupées sous le terme de dysgénésies du segment antérieur, et concernent la cornée, l'iris, l'angle irido-cornéen et le cristallin. Ce sont les syndromes de Peters, Axenfeld et Rieger, et l'embryotoxon postérieur. Des mutations des gčnes PAX6 (en 11p13), FOXC1, PITX2 et CYP1B1 ont été décrites dans le syndrome de Peters.
Les gčnes PAX6, FOXC1 et PITX2 sont des gčnes homéobox impliqués dans le développement du segment antérieur de l’oeil, tandis que le gčne CYP1B encode pour un cytochrome impliqué dans le métabolisme d’une des molécules impliquées dans le développement de la cornée.
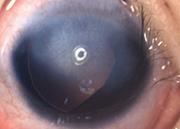
Le glaucome congénital de Peters est dű ŕ une anomalie du développement embryologique d'origine génétique lors de la migration des crętes neurales mésencéphaliques ŕ la fin de la troisičme semaine de vie intra-utérine. Cette anomalie provoque un glaucome par trabéculodysgénésie. L'atteinte est le plus souvent bilatérale. On constate une absence de développement de la membrane de Descemet et de l'endothélium au centre de la cornée entraînant une opacification de la cornée centrale avec synéchie irido-cornéenne partant de la collerette irienne et allant jusqu'ŕ l'opacité centrale. La chambre antérieure est étroite avec parfois un accolement au cristallin. Une cataracte peut ętre associée.
Les męmes cellules des crętes neurales sont responsables également de la formation du mésenchyme du cerveau antérieur, de l'hypophyse, des cartilages, des os de la partie supérieure du visage, et des papilles dentaires, ce qui explique l'association possible d'anomalies oculaires , dentaires, de troubles endocriniens et de dysmorphies faciales.
On retrouve également des hypospadias.
On a décrit :
- le syndrome de Peters type I: séparation incomplčte de la cornée et de l‘iris
- le syndrome de Peters type 2: plus sévčre, séparation incomplčte de la cornée et du cristallin , avec adhérences kératolenticulaires
- le syndrome de Peters-plus (voir ce terme) qui associe les anomalies oculaires ŕ des anomalies de l'oreille et du coeur, un nanisme, une fente palatine et un retard mental
- le syndrome de Kivlin-Krause qui associe un syndrome de Peters ŕ une petite taille.
Du point de vue ophtalmologique, on observe une opacité cornéenne blanche qui peut ętre isolée ou associée ŕ des synéchies irido-cornéennes, ŕ une cataracte ou ŕ un contact cornée-cristallin.
Le traitement du glaucome et de la cataracte, si elle est présente, doit ętre complété par une kératoplastie transfixiante en raison de l'atteinte cornéenne. On réalise souvent une greffe de cornée dans les premiers mois de vie pour préserver la vision.
Implications anesthésiques:
échocardiographie préopératoire, malvoyance, glaucome
Références :
Mise-à-jour juin 2020