(non-compaction cardiomyopathy, left ventricular hypertrabeculation)
Rare : prévalence de 0,014% dans la population des patients chez qui une échographie cardiaque est réalisée. Un diagnostic erroné de cardiomyopathie dilatée est souvent posé. Désignée comme « cardiomyopathie non classifiée » par l’OMS et cardiomyopathie primitive par l’AHA. Le myocarde a un aspect spongieux et hypertrabéculé ŕ l’échographie : l’endocarde a un aspect hypertrabéculé avec des récessus intertrabéculaires qui communiquent avec la cavité ventriculaire. Le cœur fœtal est initialement composé d’un réseau de fibres cardiomyocytaires et de trabécules où circule le sang : le processus de compaction de ce réseau progresse de l’épicarde vers l’endocarde et de la base vers l’apex durant la formation des cavités cardiaques. L’arrêt de ce processus de compaction de l’endocarde qui a normalement lieu entre la 6ème et la 8ème semaine de gestation produit cette cardiomyopathie.
Il existe des formes familiales :
- type 1 [MIM 604 169] : transmission autosomique dominante d’une mutation du gčne DTNA (18q12.1) qui code l’alpha-dystrobrévine
- type 2 [MIM 609 470] : d’une mutation d’un gčne en 11q15
- type 3 [MIM 601 493] : transmission autosomique dominante d’une mutation du gčne LDB3 (10q23.2)
- type 4 [MIM 613 424] : transmission autosomique dominante d’une mutation du gčne ACTC1 (15q14) qui code l’actine alpha cardiaque
- type 5 [MIM 613 426] : transmission autosomique dominante d’une mutation du gčne MYH7 (14q11.2) qui code la chaîne lourde 7 de la myosine
- type 6 [MIM 601 494] : transmission autosomique dominante d’une mutation du gčne TNNT2 (1q32.1) qui code la troponine cardiaque T
- type 7 [MIM 615 092] : transmission autosomique dominante d’une mutation du gčne MIB1 (18q11.2)
- type 8 [MIM 615 373] : transmission autosomique dominante d’une mutation du gčne PRDM16 (1p36.32)
- type 9 [MIM 611 878] : transmission autosomique dominante d’une mutation du gčne TPM1 (15q22.2) qui code la tropomyosine 1
- type 10 [MIM 615 396] : transmission autosomique dominante d’une mutation du gčne MYBPC3 (11p11.2)
- et une forme liée ŕ l’X (syndrome de Barth) [MIM 302 060] : mutation du gčne TAZ (Xq28) qui code la tafazzine impliquée dans le métabolisme de la cardiolipine; elle est associée ŕ une neutropénie et ŕ des anomalies mitochondriales.
Il existe des formes familiales liées à l’X (gène G4.5 en Xq28, syndrome de Barth), de transmission autosomique dominante (mutations de l’alpha-dystrobrévine ou de la protéine FKB12 qui intervient au niveau du récepteur de la ryanodine 2) ou liées à une cytopathie mitochondriale.
La présentation clinique est non spécifique : troubles du rythme ventriculaire (risque de tachycardie ventriculaire et de mort subite), syncopes, décompensation cardiaque, manifestations thromboemboliques ; parfois syndrome de Wolf-Parkinson-White chez l’enfant.
Cette cardiomyopathie peut également être associée à :
- une cardiopathie congénitale : ALCAPA, malformation d’Ebstein, cardiopathie cyanogène
- une affection métabolique ou génétique : myopathie de Duchenne ou Becker, trisomie 21, acidémie propionique, glycogénose de Pompe, syndrome de Brugada (lié ŕ une mutation du canal K4 ou Na5).
ECG : hypertrophie biventriculaire, troubles de conduction auriculo-ventriculaire, WPW.
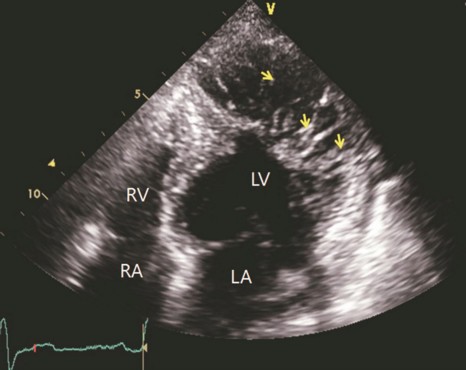
Echocardiographie : trabéculations nombreuses (>3) au niveau de l’apex et ou de la paroi libre du ventricule gauche; le myocarde présente deux couches, une large compactée et l’autre, plus fine, non-compactée : le rapport non compactée/compactée est > 2 chez l’adulte et > 1,4 chez l’enfant) (voir flčches sur l’image ci-dessous)
Traitement : symptomatique, en fonction de la clinique et de la pathologie cardiaque ou musculaire associée. Il faut parfois placer un défibrillateur interne. Chez le nourrisson, le pronostic est mauvais (< 18 mois de survie sans transplantation) si une instabilité hémodynamique, une dilatation du VG ou une mauvaise fonction ventriculaire sont présents au moment du diagnostic.
Implications anesthésiques:
rien de particulier chez les patients qui ne présentent pas de signes de décompensation cardiaque; échocardiographie préopératoire récente, éventuellement échographie transoesophagienne peropératoire s’il y a des symptômes de décompensation cardiaque ; risque de troubles du rythme ventriculaire ; tenir compte de la pathologie cardiaque ou musculaire associée; risque thrombolembolique.
Références :
- Batisse A, Fermont L, Lévy M
Cardiopathie par dysfonction myocardique.
In Cardiologie pédiatrique pratique, 4čme éd. Doin, 2013, p 187-93.
- Alehan D.
Clinical features of isolated left ventricular noncompaction in children.
Int J Cardiol 2004; 97: 233-7. - Sviggun HP, Kopp SL, Rettke SR, Rehfeldt KH.
Perioperative complications in patients with left ventricular non-compaction.
European J Anaesthesiol 2011 ; 28 : 207-12. - Zuckerman WA, Richmond ME, Singh RK, Carroll SJ, Stare TJ, Addonizio LJ.
Left-ventricular noncompaction in a pediatric population: predictors of survival.
Pediatr Cardiol 2011; 32: 406-12. - Ing RJ, Ames WA, Chambers NA.
Paediatric cardiomyopathy and anaesthesia.
Br J Anaesth 2012 ; 108 : 4-12. - Ergul Y, Nisli K, Bilge AK, Dindar A.
Aborted sudden cardiac death in a child with left ventricular non-compaction.
Pediatrics International 2013; 55: 388-91. - Kim D, Kim E, lee J-H, Kim CS, Lee SM, Lee JE.
Anesthetic experience of a patient with isolated left ventricular noncompaction.
Korean J Anesth 2016; 69: 275-9. - Kumar N, Troianos CA, Baisden JS.
Left ventricular assist device insertion in a patient with biventricular noncompaction cardiomyopathy, Ebstein anomaly, and a left atrial mass : a case report.
A&A Case Reports 2016; 7: 251-5. - Benarroch L, Bonne G, Rivier F, Hamroun D.
The 2020 version of the gene table of neuromuscular disorders.
Neuromusc Dis 2019 ; 29 : 980-1018 ou http://www.musclegenetable.fr. - Maddali MM, Eapen Thomas E, Al-Abri IA, Patel MH MD, Al-Maskari SN, Al-Yamani MI.
Dilated cardiomyopathy phenotype sssociated left ventricular noncompaction and congenital Long QT syndrome Type-2 in infancy with KCNH2 gene mutation: anesthetic considerations.
J Cardiovasc Thorac Anesth 2022 ; 36 :3662-7.
Mise-à-jour août 2022