[ MIM 251 200, 603 802, 604 317, 604 321, 604 804, 608 393, 608 716, 612 703, 614 673, 614 852, 615 414, 616 080, 616 402, 616 486, 616 681]
Rare. Prťvalence de 1/100.000 en Europe, plus frťquente au Pakistan. Trouble du dťveloppement neurologique caractťrisť par un pťrimŤtre cr‚nien rťduit ŗ la naissance sans anomalies notables de l'architecture cťrťbrale et avec un dťficit intellectuel variable.
La transmission est autosomique rťcessive. On distingue dix sous-types reposant sur base de 9 gŤnes identifiťs†: MCPH1,†WDR62,†CDK5RAP2,†CEP152,†ASPM,†CENPJ,†STIL,†CEP63,†CASC5,†PHC1†et†CEP135. Ces mutations provoquent une rťduction de la production de neurones corticaux pendant la neurogťnŤse embryonnaire. Les mutations des gŤnes CDC6,†CDT1,†ORC1,†ORC4, ORC6WDR62†peuvent provoquer des anomalies corticales plus graves. Des mutations d'un de ces gŤnes peuvent produire le syndrome de Meier-Gorlin (voir ce terme).† La MCPH et le syndrome de Seckel (voir ce terme) appartiennent ŗ un mÍme spectre clinique, les mutations (CENPJ,†CEP152) rťsultent dans l'un ou l'autre des phťnotypes.
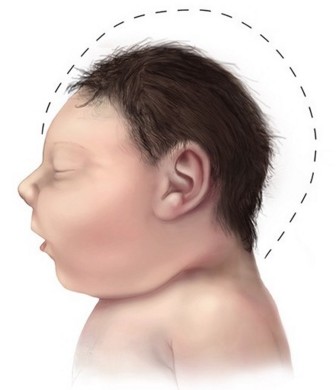
Le pťrimŤtre cr‚nien (PC) infťrieur ŗ 2 dťviations standards (DS) ŗ la naissance pour l'‚ge, le sexe et l'ethnie. La microcťphalie peut Ítre dťtectťe dŤs la 32e semaine de grossesse. La croissance cr‚nienne est ensuite trŤs lente, et le PC est rťduit : en-dessous de 3 DS avant 6 mois, et gťnťralement entre 4 et 12 DS chez les adultes. On observe un dťficit intellectuel non-progressif lťger ou modťrť, sans dťficit neurologique significatif, et parfois une ťpilepsie (10%). Un retard dans la prise prťcoce de repŤres moteurs et la parole sont courants. La plupart des patients sont hyperactifs.
Implications anesthésiques:
risque d’intubation difficile si micrognathie†; la croissance laryngotrachťale n’est pas affectťe par le microcťphalie†: choisir la sonde endotrachťale en fonction de l’‚ge.
Références :
- Nishida T, Mihara T, Horiki T, Ka K.
Anaesthesia and orphan disease†: primary autosomal recessive microcepaly-10 caused by a mutation of NF335 gene.
Eur J Anaesthesiol 2016; 33: 543-4
Mise à jour août 2016