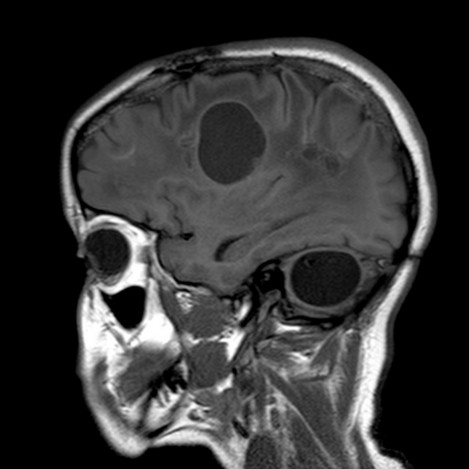
Trčs rare. Transmission autosomique récessive d’une mutation du gčne SNORD118 (17p13.1). Début variable, de l’enfance ŕ l’âge adulte. Développement progressif d’une leuco-encéphalopathie avec calcifications cérébrales et formation de kystes intracérébraux qui entraînent un déclin cognitif, des dyskinésies, une spasticité, parfois des convulsions ou une hydrocéphalie. La cause en serait une microangiopathie diffuse des vaisseaux cérébraux.
La présentation clinique est proche du syndrome de microangiopathie cérébro-rétinienne avec calcifications et kystes (CRMCC) [MIM 612 199] ou syndrome de Coats plus (voir ce terme) causé par une mutation du gčne CTC1 et oů on retrouve une ostéopénie, un risque important d’hémorragie digestive, une anémie avec thrombopénie et des anomalies de la peau et des phančres.
Implications anesthésiques:
troubles neurologiques, risque d’hypertension intracrânienne (un cas de syndrome malin aux neuroleptiques ou sérotoninergique aprčs l’administration de métoclopramide)
Références :
- Shtaya A, Elmslie F, Crow Y, Hettige S.
Leukoencephalopathy, intracranial calcifications, cysts, and SNORD188 mutation (Labrune syndrome) with obstructive hydrocephalus.
World Neurosurg 2019 ; 125 :271-2.
- Holland EL, Sanetto RP, Knipper EK.
Hypermetabolic syndrome and dyskinesia after neurologic surgery for Labrune syndrome : a case report.
A&A Practice 2020 ; 14 : e01212.
Mise-à-jour mai 2020