Une hypertension artérielle pulmonaire (dite porto-pulmonaire : HPoP) se développe chez environ 2% des patients qui présentent une hypertension portale : cette atteinte vasculaire pulmonaire serait due au passage par la circulation pulmonaire de substances toxiques d’origine splanchnique (associée peut-être à une activité résiduelle d’enzymes pancréatiques) qui échappent au « filtre hépatique ». Ces substances entraîneraient une angiogenèse pulmonaire anormale et le développement progressif d’une fibrose intimale, de lésions plexiformes spécifiques et de thromboses (organisées et recanalisées) au niveau des artères pulmonaires distales. Des cas d’hypertension artérielle pulmonaire consécutive à un shunt porto-systémique congénital et sans hypertension portale ont été décrits. Les critères diagnostiques d’HPoP sont une PaP moyenne > 25 mmHg au repos avec des résistances pulmonaires élevées (> 240 dynes.s.cm5, ou > 3 unités Wood, au contraire de l’HP liée à une circulation hyperdynamique capillaire) et une pression d’occlusion basse (au contraire d’une situation d’hypervolémie). L’HPoP est dite modérée quand la PaP moyenne est > 35 mmHg et sévère si elle est > à 45 mmHg.
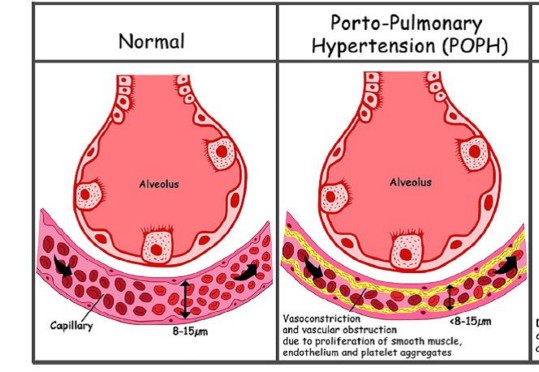
Les symptômes en sont : l’apparition d’une dyspnée d’effort, des palpitations, des syncopes. Le diagnostic est posé par l’échographie cardiaque transthoracique (Doppler) qui doit être réalisée régulièrement chez tout patient porteur d’une hypertension portale ou de shunts porto-systémiques congénitaux. Les traitements proposés sont la prostacycline, le bosentan (mais risque de toxicité hépatique), le sildénafil. La présence d’une HPoP n’est pas une indication de transplantation hépatique (dont la mortalité périopératoire dans ces cas est majeure).
Implications anesthésiques:
La prise en charge anesthésique d’un enfant souffrant d’une hypertension porto-pulmonaire implique :
- la vérification de la fonction hépatique et de l’hémostase (hypoplaquettose ?)
- l‘utilisation d’une échographie cardiaque peropératoire (balance risque/bénéfice de l’utilisation d’une sonde transoesophagienne en présence de varices oesophagiennes), ou d’une mesure directe des pressions artérielles pulmonaires ( cathéter de Swan-Ganz) indispensable pour poser le diagnostic précoce d’hypertension artérielle pulmonaire
Références :
- Chabot F, Gomez E, Boyer L, Kheir A, Le Pavec J, Sitbon O, Herve P.
Hypertension portopulmonaire.
Rev Mal Resp 2006 ; 23 : 629-41. - Ersch J, Bäzinger O, Braegger C, Arbenz U, Stallmach T.
An infant with pulmonary hypertension due to congenital porto-caval shunt.
Eur J Pediatr 2002; 161:660-2.
- Karrer FM, Wallace BJ, Estrada AE.
Late complications of biliary atresia: hepatopulmonary syndrome and portopulmonary hypertension.
Pediatr Surg Int 2017; 33: 1335-40. - Lee WS, Wong SY, Ivy D, Sokol RJ.
Hepatopulmonary syndrome and portopulmonary hypertension in children: recent advances in diagnosis and management.
J Pediatr 2018; 196; e14-21.
Mise-à-jour mai 2018