(Syndrome adrénogénital)
Groupe de 7 maladies de transmission autosomique rķcessive dues Ó des mutations de gĶnes codant pour des enzymes impliquķs dans la biosynthĶse du cortisol. La plupart des patients sont porteurs de mutations hķtķrozygotes (une mutation diffķrente sur chaque allĶle du gĶne), ce qui explique la variabilitķ des prķsentations cliniques, qui sont le rķsultat de la prķsence de l’enzyme la plus fonctionnelle. La forme la plus frķquente (90 % des cas environĀ; prķvalence 1/14.000) est le dķficit en 21-hydroxylase (mutation du gĶne CYP21A2 en 6p21.3), responsable responsable de la transformation de la progestķrone en dķsoxycorticostķrone. La forme dite classique de cette pathologie s'exprime cliniquement, dĶs la naissance dans la plupart des cas, sous 2 formes:
- une forme se traduisant par des signes de virilisation (25% des cas), plus facile à reconnaître chez la fille que chez le garçon
- une forme se traduisant par un syndrome de perte de sel et compromettant le pronostic vital en l'absence de diagnostic et de traitement précoce.
Dans les formes dites non-classiques (prķvalence estimķe de 1/200 Ó 1/1000), le dķficit est incomplet et l'anomalie enzymatique ne s'exprimera cliniquement qu'au moment de la pubertķ par des signe de virilisation (lÓ encore plus facilement identifiables chez la fille que chez le garńon). Certaines formes sont trĶs peu symptomatiques mais sont la cause de certaines formes d’infertilitķ.
Les autres dķficits enzymatiques constitutionnels qui peuvent provoquer une hyperplasie congķnitale des surrķnales sont listķs dans les deux tableaux. Le dķficit en StAR (acronyme de Steroidogenic Acute Regulatory protein) est responsable d'une forme clinique sķvĶre de l'affection dķnommķe hyperplasie congķnitale lipo’de des surrķnales [MIM 201 710].
Dans environ 10% des cas, l‘hyperplasie congķnitale des surrķnales classique est associķe Ó une forme hypermobile du syndrome d’Ehlers-Danlos: il s’agit du syndrome CAH-X , un syndrome de gĶnes contigus par dķlķtion des gĶnes CYP21A2 et TNXM (qui encode la tķnascine X) en 6p21.(voir ce terme)
.jpg)
|
dķficit |
21-hydroxylase |
11Ō-hydroxylase |
17ß-hydroxylase ou 17,20-lyase |
3Ō-hydroxy steroid dķshydrogķnase |
P450 oxydo-
|
Hyperplasie lipo’de |
Enzyme clivant la chaŅne latķrale du cholestķrol |
|
gĶne |
CYP21A2 |
CYP11B1 |
CYP17A1 |
HSD3B2 |
POR |
StAR |
CYP11A1 |
|
locus |
6p21.3 |
8q24.3 |
10q24.32 |
1p12 |
7q11.23 |
8p11.23 |
15q24.1 |
|
MIM |
|||||||
|
incidence |
1/10.000 |
1/100.000
|
1/50.000
|
rarissime |
trĶs rare |
trĶs rare (surtout Japon, Corķe, Palestine) |
rarissime
|
|
perte de sel |
oui si classique |
non |
non |
oui |
non |
oui si classique |
oui si classique |
|
hypertension |
non |
oui si classique |
oui |
non |
oui |
non |
non |
|
virilisation |
oui si classique |
oui si classique |
non |
non |
non |
non |
non |
|
dķficit hormones stķro’des sexuelles |
non |
non |
oui |
oui si classique |
oui |
oui
|
oui
|
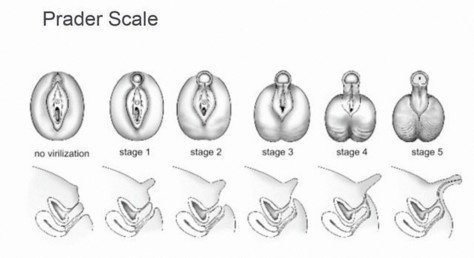
Les 5 stades de virilisation des organes gķnitaux externes chez la fille selon Prader.
L'hyperplasie congķnitale des surrķnales est dķpistķe de maniĶre systķmatique Ó 3 jours de vie par le dosage radio-immunologique de la 17 OH-progestķrone sur le test de ce qui permet d'en faire le diagnostic avant l'apparition du syndrome de perte de sel qui survient habituellement vers 2-3 semaines de vie.
Le traitement dépend des dosages biologiques. Il repose sur la compensation du déficit en glucocorticoïdes par de l'hydrocortisone et, si c'est le cas, du déficit en aldostérone (pour réduire les concentrations plasmatique d'angiotensine II). Il faut également administrer des inhibiteurs de l'aromatase (pour ralentir la maturation squelettique) et des anti-androgènes (cyprotérone, flutamide) pour abaisser les taux d'androgènes et éviter la virilisation des fillettes. En cas de dķficit en 21-hydroxylase, des ķtudes rķcentes ont montrķ l’efficacitķ de l’administration orale (2x/jour) de crinecerfont, un antagoniste du rķcepter du corticotrope releasing factor type 1, permet de diminuer les taux sanguins d’adrostĶnedione et donc leurs effets secondaires.
Implications anesthésiques:
- en période néonatale, devant un syndrome de perte de sel avec dķshydratation sķvĶre, apparu avant que le diagnostic de la maladie n'ait ķtķ posķ ;
- dosage des ķlectrolytes sanguins, de la glycķmie et de la 17-OH-progestķrone, de la rķnine, du cortisol et de l'aldostķrone car l'hypothĶse du diagnostic d'hyperplasie congķnitale devrait Ļtre systķmatiquement ķvoquķe devant une dķshydratation inattendue en pķriode nķonatale, surtout chez une fillette prķsentant des signes de virilisation ;
- rķhydratation hydroķlectrolytique par bolus de 20 mL/kg de NaCl 0,9 % en 1 heure, rķpķtķ au besoin jusqu'Ó restauration d'un ķtat hķmodynamique satisfaisant. Les enfants prķsentant un dķficit en 21-hydroxylase sont habituellement hyperkaliķmiques : il ne faut pas leur apporter de K+, et donc ķviter le lactate Ringer ; seuls ceux atteints d'un dķficit en 11Ō-hydroxylase ou en 17ß-hydroxylase peuvent Ļtre hypokaliķmiques et nķcessiter un apport de K+ : il faut attendre les rķsultats de l'ionogramme avant d'envisager d'administrer un solutķ contenant du K+;
- administration de glucose si l'enfant est hypoglycémique et, de toutes façon, dans la perfusion de base qui sera poursuivie après la restauration de la volémie.
- dosage habituel des corticoïdes et minéralocorticoïdes en cas de déficit en 21hydroxylase :
hydrocortisone p os : 10-15 mg/m2/jour en 3 prises
fludrocortisone p os : 0,05 à 0,2 mg/jour en 2 prises
suppléments de NaCl 1 à 2 g/jour (dans la petite enfance)
Ces dosages sont adaptés en fonction des contrôles biologiques et de la croissance de l’enfant (équilibre plus difficile lors de la puberté) : chez l’adulte, 15 à 25 mg/jour en 2 ou 3 prises
- en cas de fièvre, de gastroentérite avec déshydratation, de traumatisme majeur : apport de glucose et d’électrolytes en IV et hydrocortisone IV à la dose initiale de
25 mg chez le petit enfant
50 mg chez l’enfant d’âge scolaire
100 mg chez l’adulte
Les doses suivantes sont calculées sur base de «3 à 4x la dose quotidienne habituelle divisée en 4 doses/jour jusqu’au retour à une situation normale.
- lors d'une anesthésie, soit pour le traitement chirurgical de la virilisation des organes génitaux externes d'une fillette, soit pour le traitement d'une pathologie chirurgicale intercurrente (dans les deux sexes) :
Recommandations de la Haute Autorité de Santé (France), avril 2011
* en cas de décompensation aiguë (fatigabilité excessive, nausées, douleurs abdominales, hypotension, pâleur, sueurs): hydrocortisone 2 mg/kg toutes les 4-6 heures en IV et réanimation hydroélectrolytique
* en cas de chirurgie majeure:
- hospitalisation la veille de l'intervention et installation d'une voie veineuse avec apport des besoins hydriques, glucidiques et électrolytiques de base ; la dose habituelle de corticoïdes est doublée la veille de l’intervention ;
- administration de doses "de stress" d'hydrocortisone à l'induction de l'anesthésie (2 mg/kg ou 50-100 mg/m2), répétées toutes les 4-6 heures jusqu'à la reprise de l'alimentation et du traitement habituel ;
- il n'existe pas de forme injectable de minéralocorticoïdes ; pour les patients ayant un déficit en aldostérone et qui peuvent s'alimenter, il est utile de leur faire prendre une dose orale de fludrocortisone (50 à 200 µg) 2-3 heures avant l'intervention.
* en cas de chirurgie mineure ou examen nécessitant une période de jeûne : administration de doses "de stress" d'hydrocortisone à l'induction de l'anesthésie (2 mg/kg ou 50-100 mg/m2), répétées toutes les 4-6 heures jusqu'à la reprise de l'alimentation.
NB : le principe de doubler la dose de corticostéroïdes la veille de l’opération ne repose sur aucune évidence et n’est n’est recommandķ ni par The Endocrine Society, ni l’APAGBI, ni par The Royal College of Physicians, ni par The Society for Endocrinology UK. Il est probablement prķfķrable d’administrer le traitement habituel le matin de l’intervention et d’injecter une faible dose d’hydrocortisone (2 mg/kg) lors de l’induction de l’anesthķsie suivie deĀ:
* chirurgie majeureĀ: infusion continue de 25mg/24h si < 10 kg
50 mg/24h si 11-20 kg
100 mg/24h si > 20 kg et prķpubĶre
150 mg/24h si > 20kg et pubĶre
ou 2 mg/kg/4h
* chirurgie mineureĀ: doubler la dose orale lors de la rķalimentation et durant 24h
|
|
Dose équivalente (mg) |
Puissance relative |
|
hydrocortisone |
20 |
1 |
|
prednisolone |
5 |
4 |
|
méthylprednisolone |
4 |
5 |
|
dexamétasone |
0,75 |
25 |
Tableau d’équivalence des stéroïdes
Références:
- White PC, Speiser PH.
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency
.
Endocrine Reviews 2000; 21:245-291.
- Lambert SM, Vilain EJ, Kolon TF.
A practical approach to ambiguous genitalia in the newborn period.
Urol Clin North Am 2010; 37:195-205.
- Ruppen W, Hagenbuch N, Jöhr M, Christen P.
Cardiac arrest in an infant with congenital adrenal hyperplasia.
Acta Anaesthesiol Scand 2003; 47: 104-5
- Speiser PW, Azziz R, Baskin LS, Ghizzoni L et al.
Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency : an endocrine society clinical practice guideline.
J Clin Endocrinol Metab 2010; 95: 4133-60.
- El-Maouche D, Arlt W, Merke DP.
Congenital adrenal hyperplasia.
Lancet 2017; 390: 2194-2210
- Merke DP, Auchus RJ.
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
NEJM 2020; 383: 1248-61
- Woodcock T, Barker P, Daniel S, Fletcher S et al.
Guidelines for the management of glucocorticoids during the peri-operative period for patients with adrenal insufficiency.
Anaesthesia 2020;75:654-73
- Heath C, Siafarikas A, Sommerfield A, von Ungern-Sternberg BS.
Peri-operative steroid management in the paediatric population.
Acta Anaesth Scand 2021; 65:1187-94.
- Sarafoglou K, Kim MS, Lodish M, Felner EI, Martinerie L, Nokoff NJ, Clemente N, Fechner PI et al for the CAHtalyst Pediatric Trial Investigators.
Phase 3 trial of Crinecerfont in pediatric Congenital Adrenal Hyperplasia.
NEJM 2024Ā; DOI: 10.1056/NEJMoa2404655
Mise à jour juin 2024