Incidence globale entre 2 et 5/10.000 naissances vivantes. Elle est isolée dans 58 % des cas. Une origine génétique est retrouvée dans de rares cas: DIH1 (acronyme en anglais de DIaphramatic Hernia) 1 [MIM 142 340], mutation en 15q26, DIH 2 [MIM 222 400], mutation en 8p23, DIH3 [MIM 610 187], mutation du gčne ZFPM2 (8q23.1), DIH4 [MIM 617 557], mutation du gčne ALDH1A2 (15q21.3) et une forme liée ŕ l’X [MIM 306 950].
Les autres sont associées ŕ:
- des malformations cardiaques CIA, CIV, tétralogie de Fallot, coarctation de l’aorte,
- des syndromes comme le syndrome de Fryns (10%), de CHARGE, de Beckwitt-Wiedemann, de Donnai-Barrow, de Cornelia De Lange, de Denys-Drash, de Pallister-Killian, de Wolf-Hirshhorn, de Simpson-Golabi-Behmel, Kabuki, Mathew-Wood (voir ces termes)
- des anomalies chromosomiques: trisomie 13, 18 ou 21, monosomie X, certaines délétions ou duplications
- une malformation pulmonaire: une séquestration pulmonaire est présente dans environ 3,4 % des cas: le défect du diaphragme est alors souvent plus large (types C ou D) et ces patients ont plus souvent besoin d’une ECMO.
La hernie diaphragmatique résulte d’une anomalie de la fermeture du canal pleuro-péricardo-péritonéal qui permet la pénétration et le développement des viscčres abdominaux dans la cavité thoracique dčs la 8e semaine de la gestation. Dans la trčs grande majorité des cas il s’agit d’une hernie par le foramen de Bochdalek :
- en postéro-latéral gauche dans 75 ŕ 90% des cas ;
- en postéro-latéral droit dans 10% des cas environ ;
- en central (formes bilatérales) dans 1 ŕ 5% des cas (formes habituellement fatales). *
Dans les cas restants, la hernie peut se faire par le foramen de Morgagni (ou fente de Larrey) en rétrosternal (23 %) ou en central (1-2 %) des cas
Diagnostic : le plus souvent durant la grossesse dans les pays développés, ŕ l’occasion d’une échographie.
La gravité dépend de plusieurs facteurs :
- le stade fśtal auquel survient la herniation ;
- le degré d'hypoplasie pulmonaire en sachant que si l'atteinte pulmonaire est beaucoup plus marquée du côté de la hernie, elle est toujours bilatérale męme en cas de hernie unilatérale ;
- la herniation intrathoracique du foie (« liver up ») représente un important facteur de gravité : la survie est meilleure si le foie est intraabdominal
- les lésions associées : les formes isolées ont un meilleur pronostic les formes associées ŕ d'autres malformations ou ŕ des anomalies génétiques.
Les poumons présentent une diminution des divisions de l’arbre bronchique et de l’artčre pulmonaire, surtout du côté de la hernie : cela contribue de maničre mécanique ŕ provoquer une augmentation de la pression artérielle pulmonaire. La herniation thoracique des viscčres abdominaux affecte le développement anatomique et physiologique du cśur, dans les formes isolées comme dans les formes non isolées. Il existe souvent une hypoplasie relative du cśur gauche dont le remplissage a été défectueux suite ŕ la perturbation du flux du ductus venosus par les organes heniés. Toutefois, ces anomalies se normalisent assez rapidement aprčs la cure chirurgicale de la hernie et permettent de supporter une circulation biventriculaire. Il faut également noter que les enfants atteints de l'affection ont fréquemment une insuffisance surrénalienne qui aggrave notablement la morbidité respiratoire et rend probablement compte de leur fréquente résistance aux traitements par catécholamines.
Męme en l’absence de malformation cardiaque, le canal artériel et le foramen ovale ne sont pas anatomiquement fermés ŕ la naissance et les conséquences hémodynamiques et biologiques (hypoxémie, hypercarbie, acidose) de l’insuffisance respiratoire favorisent leur (ré)ouverture : un shunt droite-gauche peut se produire ŕ leur niveau, aggravant l’hypoxémie due ŕ l’hypoplasie pulmonaire. Cependant, il n’est pas rare qu’un shunt gauche-droit au niveau atrial (dű ŕ la diminution de compliance du VG) soit associé ŕ un shunt droite-gauche au niveau ductal (hypertension artérielle pulmonaire).
Les meilleurs indicateurs de pronostic actuellement retenus sont représentés par :
- le poids du fśtus ŕ la naissance (supérieur ou inférieur ŕ 2750 g),
- la présence d’une hernie du foie en intrathoracique,
- l’existence de malformations associées
- un indice échographique:
- la mesure du rapport entre la surface de l'aire pulmonaire et la circonférence de la tęte ; ce rapport O/E LHC (Observed/Expected Lung area to Head Circumference ratio) est exprimé en pourcentage du rapport normal attendu pour l'âge du fśtus. Selon ce %, on parle de forme extręme (< 15%), sévčre (16-25 %), modérée (26-35 %) ou légčre (36-45 %). En cas de hernie diaphragmatique gauche isolée, le pronostic vital est nul en cas de forme extręme, de 20 % en cas de forme sévčre, de 55 % pour les formes modérées et de 85 % pour les formes légčres.
Lorsque les indicateurs de pronostic sont mauvais:
- hernie gauche: rapport O/ELHC < 25 % ou entre 25 et 35 %, quelle que soit la position du foie, ou 35 ŕ 45 % avec herniation intrathoracique du foie
- hernie droite: O/ELHC < 45 %
- hernie bilatérale: O/ELHC < 25 %
certaines équipes tentent de favoriser la croissance pulmonaire en réalisant une occlusion de la trachée fśtale par voie endoscopique percutanée (« FETO » pour Fetal Endoscopic Tracheal Occlusion). Cette technique consiste ŕ introduire un ballonnet d’occlusion vasculaire dans la trachée par voie translaryngée entre la 26čme et la 28čme semaine.
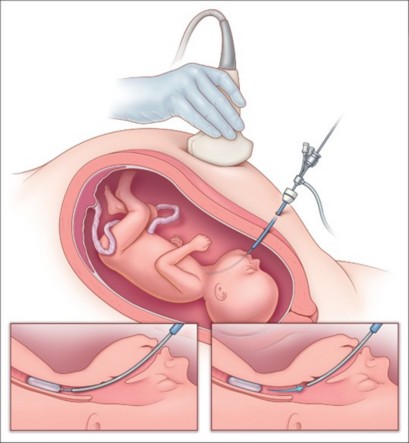
Ce ballonnet (7-8 mm de diamčtre x 20-22 mm de long) est maintenu gonflé (0,6 ml de NaCl 0,9 %) jusque vers la 34čme semaine: la pression intratrachéale diminue en effet la nombre de pneumocytes de type II et expose donc ŕ un risque de déficit en surfactant. Le retrait du ballon s’effectue par voie endoscopique percutanée ou ponctionné sous échoguidage. Cette technique permet d'obtenir un taux de survie de l'ordre de 50 % en cas de forme sévčre. La survie n’est pas améliorée en cas de forme modérée. Lors du retrait du ballonnet, le risque de naissance prématurée est multiplié par 2,6 en cas de forme sévčre et par 2,9 en cas de forme modérée.
Le diamčtre de la trachée est en moyenne augmenté de 31% chez les nouveau-nés (trachéomégalie) qui ont bénéficié d’un FETO et ils présentent un risque supplémentaire (environ x5) de trachéomalacie (hyperlaxité de la pars membranacea).
Diagnostic ŕ la naissance: lorsque le diagnostic n’est pas porté avant la naissance, la symptomatologie clinique présentée par le nouveau-né dépend du degré d'hypoplasie pulmonaire, de la masse de viscčres dans la hernie, du degré de déviation médiastinale, de la présence d'un iléus et d'une dilatation intestinale, et, finalement, de l'importance de l'hypertension artérielle pulmonaire. Dans les formes habituelles, le nouveau-né développe rapidement aprčs la naissance une tachypnée, une dyspnée, une cyanose.
A l'examen physique, on retrouve, en plus des signes de détresse respiratoire :
- une diminution ou une absence de murmure vésiculaire ;
- du tympanisme au niveau de l'hémithorax sičge de la hernie ;
- et un abdomen déprimé en creux, dit « scaphoďde ».
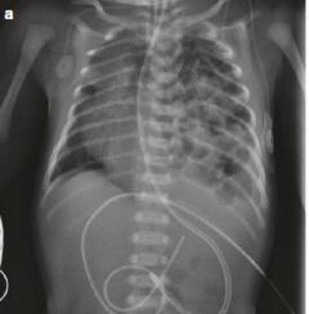
Les bruits cardiaques sont assourdis et déplacés du côté opposé ŕ la hernie. On peut parfois déceler du péristaltisme au niveau thoracique. La radiographie thoracique montre des anses intestinales dans l'hémithorax, une déviation controlatérale du médiastin, un aspect médiocre du tissu pulmonaire et, parfois, un bouclage intra-thoracique de la sonde gastrique (enroulée dans un estomac en partie ou en totalité situé dans le thorax).
Face ŕ la détresse respiratoire, la conduite initiale est l'assistance ventilatoire avec intubation d'emblée (si possible sans ventilation au masque facial en pression positive pour éviter d'insuffler l'estomac en position intrathoracique), l'oxygénation et la décompression gastrique. La suite de la prise en charge est la męme que dans les formes diagnostiquées avant la naissance (voir plus loin).
Il existe également des formes peu symptomatiques, ŕ révélation tardive, associées ŕ une hypoplasie pulmonaire minime et qui sont découvertes ŕ l’occasion d’un épisode de dyspnée (lors d’une infection respiratoire par exemple) ou par hasard, lors d’une radiographie de thorax. Il s’agit le plus souvent des hernies antérieures par le foramen de Morgagni mais d’authentiques hernies par le foramen de Bochdalek peuvent ne se démasquer que beaucoup plus tard dans la vie.
Prise en charge programmée : dans un centre tertiaire.
Les critčres pronostiques sont réguličrement réévalués et des interventions prénatales sont éventuellement réalisées (voir plus haut). La naissance est généralement planifiée vers la 36-38čme semaine en présence de l’équipe des intensivistes néonataux qui vont prendre en charge l’enfant. Le nouveau-né est intubé immédiatement aprčs la naissance (souvent avant de clamper le cordon ombilical) , analgésié, curarisé et transféré ŕ l'unité de réanimation néonatale oů il va ętre placé sous ventilation conventionnelle peu agressive (PIP <25 cmH2O) avec une fréquence rapide (40-60/min) (pour limiter le barotraumatisme pulmonaire et les risques de pneumothorax) et en tolérant une hypercapnie permissive (paCO2 50-70 mmHg). L’objectif visé est d’améliorer l’oxygénation (FiO2 titrée pour obtenir une SpO2 préductale entre 80 et 95%), de corriger les déséquilibres acido-basiques, de réduire le shunt droite-gauche et d’améliorer sa perfusion pulmonaire en contrôlant notamment l‘hypertension artérielle pulmonaire. En cas d'échec de la ventilation conventionnelle ou d’emblée dans certaines équipes, on recourt ŕ la ventilation par oscillation (pression moyenne 14-16 cmH2O, ÄP 30-40 cmH2O), en introduisant du NO si la SpO2 préductale est basse suite ŕ un shunt droit-gauche au niveau atrial (ŕ éviter en cas de shunt gauche-droit au niveau atrial !), avec l’objectif d'augmenter la perfusion pulmonaire ainsi que l'oxygénation. En cas de dysfonction du VG, la milrinone peut ętre utile. Il existe souvent une période d'amélioration relative et de stabilisation de l'enfant ("lune de miel ") durant les 24 premičres heures avant une possible détérioration secondaire, parfois létale.
Selon les équipes, on procčde soit ŕ une chirurgie précoce en laissant délibérément le canal artériel ouvert (perfusion de prostacycline) qui sert alors de soupape de décharge en cas de crise d’HTAP, soit on diffčre la chirurgie de quelques jours jusqu’ŕ la stabilisation hémodynamique et respiratoire de l’enfant, pour permettre le développement d’une certaine maturation pulmonaire préopératoire et une diminution de l’hypertension artérielle pulmonaire.
Les critčres d’opérabilité recommandés en Europe sont :
- une PAM normale pour l’âge (45 mmHg)
- une SpO2 préductale de 85% ŕ 95% avec une FiO2 < 50%
- un taux de lactates sanguins < 3 mmol/L
- un débit urinaire > 1 ml/kg/h
Si l’état de l’enfant ne se stabilise pas ou, a fortiori s’il se détériore, de nombreuses équipes envisagent alors, aprčs un délai variable, le recours ŕ l'oxygénateur ŕ membrane (ECMO) dans les institutions qui en disposent. Les critčres de mise en place d’une ECMO reposent habituellement sur :
- une PaCO2> 67 mmHg malgré une ventilation optimale (PIP > 28 cm H2O en ventilation conventionnelle)
- des SpO2 pré-ductales ≤ 80 % ou post-ductales ≤ 70 %
- un PH < 7,15 et un taux de lactates > 5 mmol/l
- une hypotension systémique réfractaire malgré un remplissage et un support hémodynamique avec des inotropes, et un débit urinaire < 0,5 ml/kg/h
- l’impossibilité d’abaisser l’index d'oxygénation ŕ moins de 30 (index d’oxygénation = pression de ventilation moyenne x FiO2/PaO2 x 100),
Chirurgie ouverte classique
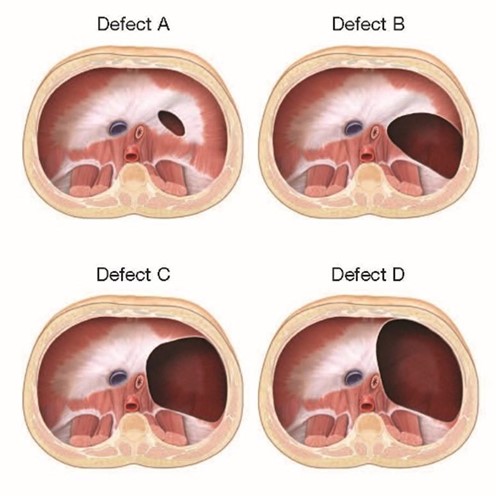
Incision abdominale sous-costale du côté atteint (gauche le plus souvent) et réintégration des viscčres herniés dans la cavité abdominale suivie de la réparation du diaphragme: en fonction de la taille du défect, le diaphragme est réparé soit directement (défects A ou B) soit ŕ l'aide d'un patch de Goretex ( défects C ou D). Le taux de récidive en cas de patch (quel que soit le matériau utilisé) est élevé (46 %).
|
Classification internationale des hernies diaphragmatiques A: petit défect uniquement musculaire B: 50-75% du diaphragme est présent, le défect inclut < 50% de la paroi thoracique C: 25% du diaphragme est présent, le défect inclut > 50% de la paroi thoracique D: peu ou pas de diaphragme (agénésie) |
Parfois, l'importance du contenu abdominal intrathoracique nécessite un double abord, thoracique et abdominal. La cavité abdominale est hypoplasique et peut rendre la fermeture difficile : il n’est pas rare de devoir placer transitoirement une prothčse pariétale en silastic de maničre ŕ éviter :
- une hyperpression intra-abdominale (que l’on peut estimer en mesurant la pression intra-gastrique ou intra-vésicale, toutes deux étant normalement inférieures ŕ 20 cm H2O) avec tous les risques d’un syndrome de compartiment abdominal (ischémie intestinale, insuffisance rénale),
- et une diminution de la compliance thoracique.
On place souvent un drain thoracique non-aspiratif du côté opéré pour permettre le drainage de l’air et de sécrétions sans provoquer de déplacement médiastinal.
La complexité de la prise en charge anesthésique dépend de l’état de stabilisation hémodynamique (pression artérielle pulmonaire supérieure ou inférieure ŕ la pression systémique) et des modalités de ventilation de l’enfant en préopératoire immédiat. Le transfert de l’enfant en salle d’opération est un temps délicat. En principe, l’enfant arrive avec une sonde naso-trachéale en place et il est ventilé, soit mécaniquement soit, plus souvent, manuellement pendant toute la phase de transfert.
S’il était sous ventilation conventionnelle, celle-ci sera poursuivie en peropératoire en visant ŕ maintenir les valeurs de PaO2 et de PaCO2 préopératoires avec, idéalement, une saturation artérielle supérieure ŕ 90% (ou une PaO2> 60 mmHg), en tentant d’éviter tout épisode d’hypercapnie majeure pour ne pas augmenter fortement les résistances pulmonaires et aggraver le shunt droit-gauche.
On peut entretenir l’anesthésie avec :
- une faible concentration de sévoflurane
- associée ŕ un morphinique (sufentanil) administré par titration
- une curarisation profonde par un agent non dépolarisant
Si l’enfant est sous NO, il faut en continuer l'administration peropératoire. Certaines équipes recourent ŕ l’utilisation d’un injecteur spécifique (INOvent®) en administrant un débit de gaz frais égal ou supérieur ŕ la ventilation-minute du patient ; s’il est utilisé avec un circuit-filtre, cet injecteur doit ętre placé sur la branche inspiratoire, en aval de la valve inspiratoire et de l’absorbeur de CO2. La plupart des équipes cependant utilise le respirateur sur lequel était branché l’enfant en réanimation néonatale, ce qui impose une technique d’anesthésie intraveineuse totale, les évaporateurs d’halogénés ne pouvant ętre connectés ŕ ce type de respirateur. Il faut contrôler les PaCO2 de façon trčs rapprochée du fait des fortes variations de compliance thoracique pendant la phase de réduction de la hernie et du risque d'hypoperfusion cérébrale en cas d’hypocapnie sévčre.
Si l’enfant est sous ECMO, les difficultés sont essentiellement liées ŕ l’anticoagulation.
La correction chirurgicale des formes peu symptomatiques ŕ révélation tardive est identique mais la prise en charge anesthésique est plus simple car il n’y a pas de problčme de ventilation ni d’oxygénation. L'usage du N2O reste contre-indiqué et, en plus du monitorage habituel, il est utile de poser une ligne artérielle pour la mesure de pression artérielle invasive et pour pouvoir surveiller réguličrement la PaO2 et la PaCO2. En cas de distension gastrique, il est impératif de décomprimer l’estomac avant l’induction d’anesthésie ou immédiatement aprčs l’intubation (éventuellement par un drainage transpariétal ŕ l’aiguille) pour éviter une compression du cśur et des gros vaisseaux qui peut entraîner un choc voire un arręt cardiaque. Dans ces formes oů l’enfant peut ętre mobilisé sans conséquences hémodynamiques importantes, il est intéressant d’assurer l’analgésie par voie épidurale si le bilan de coagulation est normal car cette technique assure une excellente analgésie per et postopératoire et limite le nombre des poussées d’hypertension artérielle pulmonaire
Les indications de chirurgie dite « minimalement invasives », par laparoscopie principalement, thoracoscopie ou l’association des 2 techniques (souvent par conversion d’une laparoscopie en thoracoscopie) ne cessent de s’élargir. La plupart des équipes attendent 1-3 jours avant d’opérer et que la pression artérielle systolique pulmonaire (estimée par échographie) soit inférieure ŕ 2/3 de la pression artérielle systolique systémique.
En pratique la chirurgie laparoscopique donne de bons résultats chez les enfants dont l’état respiratoire n’est pas trop grave et sa morbidité est alors inférieure ŕ celle de la chirurgie ouverte. Dans une série multicentrique de 151 cas opérés sous thoracoscopie, le taux de récidive précoce était plus important qu’en cas de chirurgie ouverte, surtout s’il avait fallu placer un patch pour fermer le défect diaphragmatique. La prise en charge anesthésique d’une laparoscopie ne diffčre pas sensiblement de celle de la chirurgie ouverte ŕ la différence de l’hypercapnie plus importante (avec acidose respiratoire), des épisodes plus fréquents d’hypoxémie cérébrale et de la durée souvent plus longue des interventions (risque d’hypothermie).
Suites opératoires et devenir ultérieur
En période postopératoire, on constate souvent une détérioration transitoire de la ventilation due ŕ la diminution postopératoire de la compliance thoracique. Malgré l'amélioration de la prise en charge, la mortalité des enfants atteints de hernie diaphragmatique reste élevée, autour de 20% pour les cas favorables dans les meilleurs centres, mais atteignant ou dépassant 50% dans les formes sévčres, avec malformations associées et/ou requérant un traitement par ECMO. Le taux de récidive nécessitant réintervention est de l’ordre de 10% en cas de réparation primaire du diaphragme mais de 50% si un patch (surtout s’il s’agit d’un patch résorbable) a été nécessaire.
Les autres complications ŕ distance les plus fréquentes sont :
- des déformations thoraciques (46%), plus fréquentes aprčs une forte prématurité, la mise en place d’un patch, une hernie gastrique intra-thoracique, une ventilation post-chirurgicale prolongée, un besoins d’O2 au long cours ;
- une obstruction intestinale (13%), lŕ encore plus fréquente lorsqu’un patch a été nécessaire ;
- une scoliose (13%) ;
- des problčmes respiratoires chroniques : bronchodysplasie, hypertension artérielle pulmonaire (traitement par sildénafil)
- pour ce qui concerne l‘hypertension artérielle pulmonaire:
- certains enfants n’en présentent aucune et ont un excellent pronostic;
- d’autres présentent une hypertension pulmonaire qui se corrige en 4-6 semaines (traitement par sildénafil);
- d’autres gardent une hypertension pulmonaire quasi systémique: ils nécessitent un cathétersime cardiaque pour diagnostiquer une anomalie des artčres pulmonaires, des veines pulmonaires ou du VG: en cas d’anomalie du coeur gauche, ils bénéficient parfois de l’addition de milrinone ou d’un inhibiteur de l’enzyme de conversion; sinon, il faut renforcer le traitement vasodilateur pulmonaire; leur pronostic est sombre.
Un nombre important d’enfants opérés de hernie diaphragmatique nécessite la réalisation ultérieure d’une fundoplication. De plus, on constate une diminution progressive du rapport ventilation/perfusion avec la croissance (1,58 ŕ 1 an đ 1,82 ŕ 15 ans): elle est due ŕ une diminution de la perfusion du côté hernié et est plus importante aprčs la réparation d’un défect important (C ou D).
Implications anesthésiques:
Implications anesthésiques : chirurgie néonatale, SpO2 pré- et post-ductale, ligne artérielle, monitorage de l’oxygénation cérébrale ; difficultés de ventilation ; support hémodynamique en cas d’hypotension ; dopamine ou noradrénaline, parfois dobutamine. Echocardiographie préopératoire : pression artérielle pulmonaire (shift du septum interventriculaire, shunt Dr-G au niveau du canal artériel), sens du shunt au niveau du foramen ovale, cardiopathie associée, fonction du VG ? L'hyperréactivité vasculaire pulmonaire est imprévisible et peut se révéler brutalement. Mesure de la pression intragastrique ou intravésicale en cas de difficulté de la fermeture de l’abdomen.
Références :
- Lally KP, Lasky RE, Lally P, Bagolan P et al.
Standardized reporting for congenital diaphragmatic hernia - an international consensus.
J Pediatr Surg 2013; 48:2408-15.
- Doné E, Gucciardo L, Van Mieghem T, Jani J, Cannie M, Van Schoubroeck D, Devlieger R, Catte LD, Klaritsch P, Mayer S, Beck V, Debeer A, Gratacos E, Nicolaides K, Deprest J.
Prenatal diagnosis, prediction of outcome and in utero therapy of isolated congenital diaphragmatic hernia.
Prenat Diagn 2008; 28:581-91.
- Vogel M, McElhinney DB, Marcus E, Morash D, Jennings RW, Tworetzky W.
Significance and outcome of left heart hypoplasia in fetal congenital diaphragmatic hernia.
Ultrasound Obstet Gynecol 2010; 35:310-7.
- Kamath BD, Fashaw L, Kinsella JP.
Adrenal insufficiency in newborns with congenital diaphragmatic hernia.
J Pediatr 2010; 156:495-7.
- Hirose S, Farmer DL, Lee H, Nobuhara KK, Harrison MR.
The ex utero intrapartum treatment procedure: Looking back at the EXIT.
J Pediatr Surg 2004; 39:375-80.
- Keller RL, Hawgood S, Neuhaus JM, Farmer DL, Lee H, Albanese CT, Harrison MR, Kitterman JA.
Infant pulmonary function in a randomized trial of fetal tracheal occlusion for severe congenital diaphragmatic hernia.
Pediatr Res 2004; 56:818-25.
- Harrison MR, Keller RL, Hawgood SB, Kitterman JA, Sandberg PL, Farmer DL, Lee H, Filly RA, Farrell JA, Albanese CT.
A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia.
N Engl J Med 2003; 349:1916-24.
- Bösenberg AT, Brown RA.
Management of congenital diaphragmatic hernia.
Curr Opin Anaesthesiol 2008; 21:323-31.
- Congenital Diaphragmatic Hernia Study Group, Bryner BS, West BT, Hirschl RB, Drongowski RA, Lally KP, Lally P, Mychaliska GB.
Congenital diaphragmatic hernia requiring extracorporeal membrane oxygenation: does timing of repair matter?
J Pediatr Surg 2009; 44:1165-71.
- Harting MT, Lally KP.
Surgical management of neonates with congenital diaphragmatic hernia.
Semin Pediatr Surg 2007; 16:109-14.
- Migliazza L, Bellan C, Alberti D, Auriemma A, Burgio G, Locatelli G, Colombo A.
Retrospective study of 111 cases of congenital diaphragmatic hernia treated with early high-frequency oscillatory ventilation and presurgical stabilization.
J Pediatr Surg 2007; 42:1526-32.
- Kutzsche S, Sangolt GK, Schistad O, Sunde S.
Severe complications during the management of a child with late presentation of a diaphragmatic hernia.
Acta Anaesthesiol Scand 2003; 47:1302-4.
- Gourlay DM, Cassidy LD, Sato TT, Lal DR, Arca MJ.
Beyond feasibility: a comparison of newborns undergoing thoracoscopic and open repair of congenital diaphragmatic hernias.
J Pediatr Surg 2009; 44:1702-7.
- McHoney M, Giacomello L, Nah SA, De Coppi P, Kiely EM, Curry JI, Drake DP, Eaton S, Pierro A.
Thoracoscopic repair of congenital diaphragmatic hernia: intraoperative ventilation and recurrence.
J Pediatr Surg 2010; 45:355-9.
- Deprest J, Breysem L, Gratacos E, Nicolaides K, Claus F t al.
Tracheal side effects following fetal endoscopic tracheal occlusion for severe congenital diaphragmaic hernia.
Pediatr Radiol 2010; 40: 670-3
- Snoek KG, Reiss IKM, Greenough A, Capolupo I, Urlesberer B et al.
Standardized postnatal management of infants with congenital diaphragmatic hernia in Europe: the CDH EURO Consortium Consensus -2015 Update.
Neonatology 2016; 110: 66-74.
- Quinney M, Wellespey H.
Anaesthetic management of patients with a congenital diaphragmatic hernia.
BJA Education 2018; 18: 95-101.
- Wehrmann M, Patel SS, Haxel C, Cassidy C, Howley L, Cunoe B, Gien J, Kinsella JP.
Implications of atrial-level shunting by echocardiography in newborns with congenital diaphragmatic hernia.
J Pediatr 2020; 219: 43-7
- Dao DT, Kamran A, Wilson JM, Sheils CA et al.
Longitudinal analysis of ventilation perfusion mismatch in congenital diaphragmatic hernia survivors.
J Pediatr 2020; 219:160-6
- Chatterjee D, Ing RJ, Gien J.
Update on congenital diaphragmatic hernia.
Anesth Analg 2020; 131: 808-21.
- Coughlin MA, Gupta VS, Ebanks AH, Harting MT et al.
Incidence and outcomes of patients with congenital diaphragmatic hernia and pulmonary sequestration.
J Pediatr Surg 2021; 56:1126-9.
- Deprest JA, Nicolaides KH, Benachi A, Gratacos E, Ryan G et al.
Randomized trial of fetal surgery for severe left diaphragmatic hernia.
N Engl J Med 2021; 385:107-18
- Deprest JA, Benachi A, Gratacos E, Nicolaides KH, Berg C et al.
Randomized trial of fetal surgery for moderate left diaphragmatic hernia.
N Engl J Med 2021; 385:119-29
- Basurto D, Watananirun K, Cordier A-G, Otańo J, Carriere D, Scuglia M, Moraes de Luna Freire Vargas A, Prat J, Russo FM, Debeer A, Peralta CFA, De Coppi P, Gratacós E, Benachi A, Deprest J.
Tracheomalacia and tracheomegaly in infants and children with congenital diaphragmatic hernia managed with and without fetoscopic endoluminal tracheal occlusion (FETO): a multicentre, retrospective cohort study.
Lancet Child Adolesc Health 2024 ; 8 : 580-8 doi.org/10.1016/ S2352-4642(24)00109-3
Mise à jour: septembre 2024