Acronyme en anglais de de Gastro Intestinal Stromal Tumors
Rare : incidence de 10 ŕ 15 cas par an et par million ; représente environ 1% des tumeurs du tube digestif. Pic de fréquence vers 50-60 ans. Les tumeurs stromales gastro-intestinales sont des tumeurs conjonctives, en général sporadiques, localisées généralement dans l’estomac ou le gręle. Elles dérivent des cellules de Cajal (responsables du péristaltisme) ou d’un de leurs précurseurs, et expriment les récepteurs de cytokines CD117/KIT+ (95 % des cas) et DOG-1+ (95 % des cas) ŕ leur surface. 60 % environ des GIST sičgent dans l’estomac, 30 % dans l’intestin gręle, et environ 5 % dans le côlon ou le rectum.
On distingue:
Formes dites adultes:
pic de fréquence vers 50-60 ans ; une mutation en général somatique des gčnes KIT (gręle ou colon) ou platelet derived growth factor receptor alpha (PDGFRA) (estomac) en 4q12, codant pour des récepteurs de type tyrosine kinase est retrouvée dans environ 85 % des cas. Ces mutations, induisent une activation des protéines KIT ou PDGFRA.
Formes dites pédiatriques:
surviennent chez l’enfant ou l’adulte jeune (< 30 ans), préférentiellement de sexe féminin. Elles sont souvent multiples et gastriques. Leur évolution est généralement lente, avec possibilité de métastases ganglionnaires . Elles ne présentent pas de mutation de KIT/PDGFRA, mais une perte d’expression somatique de SDH-B en immunohistochimie. Les GIST avec perte d'expression de SDH-B peuvent donner des métastases dans les ganglions lymphatiques de drainage.
Formes syndromiques:
1. neurofibromatose de type 1 : mutation du gčne NF1, sans mutation somatique des gčnes KIT et PDGFRA, GIST multiples, prédominant dans l’intestin gręle
2. triade de Carney (trčs rare) : pas de mutation constitutionnelle mais déficit de la SDH-B (sous-unité B de la succinate déshydrogénase) (gčne SDHB en 1p36.13), femme jeune, 3 entités synchrones ou métachrones : GIST gastriques multiples, chondrome pulmonaire et paragangliome extra-surrénalien
3. syndrome (ou dyade) de Carney-Stratakis (mutation constitutionnelle ou altération épigénétique de la sous-unité A, B, C ou D de la SDH) : GIST gastriques multiples, paragangliome extra-surrénalien (pas de chondrome pulmonaire)
Formes familiales:
transmission autosomique dominance avec pénétrance variable
1. d’une mutation constitutionnelle du gčne KIT (4q12)(exceptionnelle) : sujet jeune, antécédent familial de GIST, trouble de la motilité intestinale (dysphagie, constipation), tâches pigmentées cutanées, hyperplasie des cellules de Cajal,
2. d’une mutation constitutionnelle du gčne PDGFRA (4q12)[MIM 175 510]: qui entraîne le syndrome GIST plus (autrefois appelé neurofibromatose intestinale): association de GIST, des polypes inflammatoires (estomac et intestin gręle)et des tumeurs fibroďdes ŕ un facičs aux trais grossiers, des mains et pieds larges, une perte précoce des dents
3. d’une mutation du gčne SDHB (1p36.13) ou SDHC (1q23.3): cellules épithélioďdes, sujet jeune, antécédent familial de GIST.
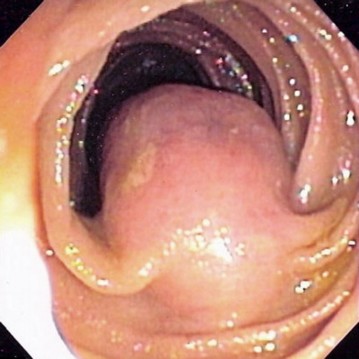
Les GIST sont souvent indolentes, elles peuvent se manifester par une hémorragie digestive, des douleurs, une anémie, une dysphagie, une occlusion ou une masse abdominale. Le risque de récidive est évalué par différentes classifications basées sur des séries rétrospectives.
Par exemple, celle de Joensuu:
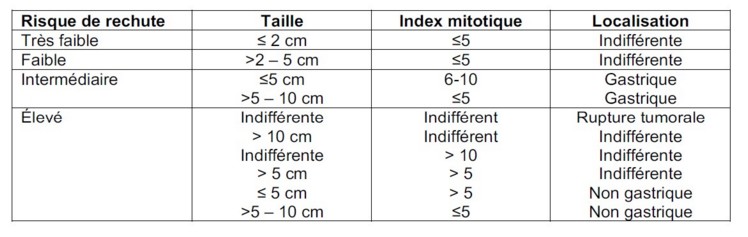
Traitement : exérčse chirurgicale complčte. Risque important de sarcomatose péritonéale en cas de rupture tumorale. En cas de métastases ou de haut risque de récidive : traitement par imatrinib (Glyvec®), un inhibiteur de la tyrosine-kinase, p os surtout en cas de mutation du gčne KIT. En cas d’échec ou de résistance ŕ l’imatrinib : sunitinib.
Implications anesthésiques:
vérifier l’absence d’anémie ; selon la localisation ; chirurgie de type oncologique ; interactions médicamenteuses de l’imatrinib.
Références :
- Landi B, Blay JY, Bonvalot S, Brasseur M, Coindre JM et al.
Thésaurus National de Cancérologie Digestive (TNCD).
Dig Liver Dis. 2019 ; 51:1223-31. doi:10.1016/j.dld.2019.07.006.
Mise à jour juillet 2020