(déficit en acétyl-CoA thiolase mitochondriale, déficit en T2, acidurie alpha-méthylacéto-acétique,MATD)
Rarissime. Cytopathie mitochondriale (voir ce terme) suite la transmission autosomique récessive d’une mutation du gène ACAT1 en 11q22.3. Il existe 6 thiolases intracellulaires (cytosol, peroxysomes, mitochondries) : cette pathologie concerne la thiolase mitochondriale et est donc aussi appelée : déficit en acétyl-CoA thiolase mitochondriale. Cette enzyme est impliquée dans le métabolisme des corps cétoniques et de l’isoleucine.
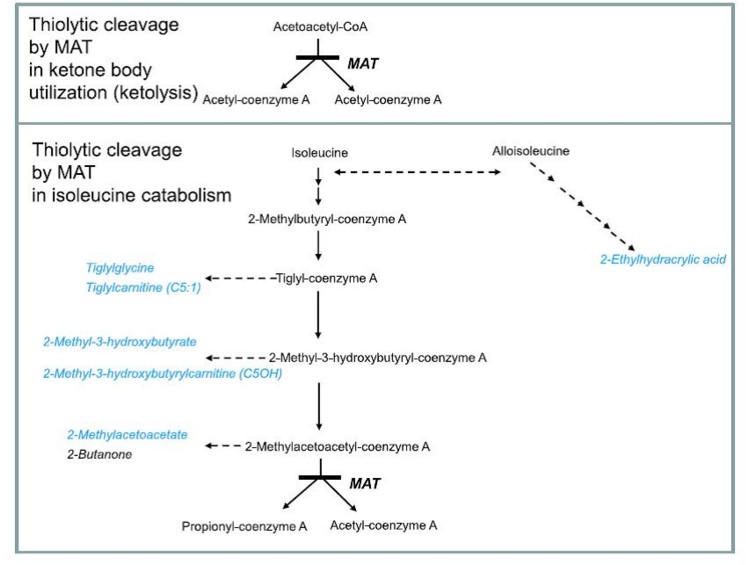
Cette affection se déclare dans l’enfance (avant dix ans, rarement en période néonatale : âge médian 12 mois) par des épisodes d’acidocétose sévčre avec ou sans hypoglycémie, parfois accompagnés de coma, en général liés au jeűne ou ŕ une infection intercurrente, plus rarement par une perte progressive des acquis intellectuels et moteurs.
IRM : lésions des noyaux gris de la base.
Traitement : régime pauvre en protéines (< 2g/kg/j), administration de glucose en cas de jeűne, suppléments de carnitine.
Le pronostic est bon si le diagnostic est précoce et le traitement bien suivi.
Implications anesthésiques:
voir cytopathies mitochondriales
Références :
- Pandey R, Singh PM, Garg R, Darlong V, Punj J.
Perioperative concerns in a beta-ketothiolase-deficient child.
J Anesth 2015 ; 29 : 647.
- Grünert SC, Sass JO.
2-methylacetoacetyl-coenzyme A thiolase (beta-ketothiolase) deficiency : one disease, two pathways.
Orphanet J Rare Diseases 2020 ; 15 : 106
Mise-à-jour: mai 2020