Aussi appelé cycle de l'ornithine, ce cycle a lieu dans des hépatocytes périportaux du foie. Il se déroule en partie dans le cytoplasme et en partie dans les mitochondries, et a pour but de transformer le NH4 en urée (schéma). Ce cycle génčre également des protons H+, ce qui explique qu'en cas d'insuffisance hépatique, l'organisme tend vers une alcalose métabolique.
Le bon fonctionnement du cycle peut ętre altéré par une anomalie d’une des 6 enzymes (voir fiches spécifiques) qui le composent, ou d’un transporteur transmembranaire (syndrome triple H).
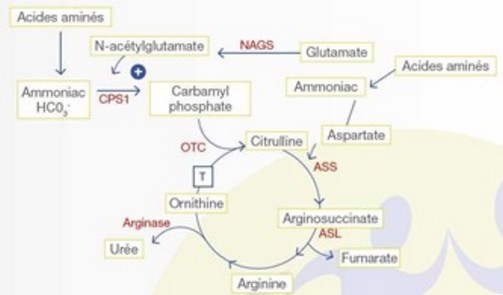
Rappel physiologique:
dans la mitochondrie,
- la carbamyl phosphate synthétase I (CPS I, 2q35), produit le carbamyl-phosphate ŕ partir de NH4+), d’ATP et de CO2. Cette réaction se fait en deux étapes consommant chacune une molécule d'ATP. La CPS I doit ętre activée par le N-acétylglutamate. Cette réaction est inhibée par la valproate, ce qui explique que le valproate (Dépakine) peut ęte ŕ l’origine d’une décompensation d’une anomalie jusque-lŕ peu symptomatique du cycle de l’urée.
- le N-acétylglutamate est synthétisé par l’association de glutamate ŕ de l’acétyl-CoA par la N-acétylglutamate synthétase (NAGS, 17q21.31)
- le carbamyl-phosphate se condense avec l'ornithine pour donner la citrulline. Cette réaction est catalysée par l'ornithine transcarbamylase (OTC, Xp21.1).
- la citrulline est exportée dans le cytoplasme par le transporteur d’ornithine et de citrulline de la membrane mitochondriale (ORNT1, 13q14). Le déficit en ORNT1 provoque le syndrome triple H (hyperornithinémie-hyperammoniémie-homocitrullinurie).
dans le cytoplasme,
- la citrulline se condense, en présence d'ATP, avec une molécule d'acide aspartique grâce ŕ l'arginosuccinate synthase (ASS, 9q34)
- l'acide arginosuccinique est converti en arginine et en acide fumarique par l'arginosuccinate lyase (ASL, 7q11.2).
- l'arginase 1 (ARG1, 6q23) catalyse l'hydrolyse de l'arginine en ornithine et urée en consommant une molécule d'eau. L'ornithine est ainsi régénérée et peut regagner la mitochondrie pour fixer une nouvelle molécule de carbamyl-phosphate avec ORNT1.
Les signes cliniques sont variables : coma métabolique, neurologiques (convulsions, signes focaux), hépatiques (hépatomégalie, cytolyse, insuffisance hépatocellulaire) ou neuropsychiatriques (ataxie, troubles du comportement). Ils varient également en fonction de l'âge d'apparition des premiers symptômes :
- période néonatale: hyperammoniémie avec vomissements, léthargie ou irritabilité ;
- nourrisson : anorexie, vomissements, retard de développement, hépatomégalie. On recherche souvent une cause digestive ŕ ces problčmes avant de poser le diagnostic ;
- enfance et adolescence : tableau d'encéphalopathie aiguë (syndrome de Reye) ŕ l'occasion d'un stress (fičvre, catabolisme protéique, postopératoire) ou de prise de valproate (Dépakine), ou signes neurologiques progressifs (retard mental, convulsions, ataxie). Une corticothérapie (augmentation du catabolisme protéique) ou une chimiothérapie ŕ base d’asparaginase peut également ętre un facteur déclenchant. Certains patients présentent un dégoűt spontané pour la viande ou les repas riches en protéines.
- le post-partum est également une période ŕ risque de décompensation ŕ cause de la réaction catabolique induite par l’involution utérine.
Principe de base du traitement : prévention de l'hyperammoniémie par un régime pauvre en protéines (la teneur en protéines est adaptée en fonction de l'âge pour tenir compte de la croissance et doit ętre diminuée en cas d'infection ou de stress important comme une chirurgie pour limiter le catabolisme protéique et la formation de NH4 ; de plus, il faut souvent ajouter du benzoate de Na (0,25 g/kg/j), du phénylbutyrate de Na (0,25 g/kg/j) qui stimulent des voies métaboliques alternatives et diminuent la synthčse de NH4, ou de l'HCl d'arginine (0,1 ŕ 0,2 g/kg/j) qui favorise la formation d’urée par d’autres voies.
Implications anesthésiques:
- contacter l’équipe qui suit habituellement l’enfant
- limiter la durée du jeűne préopératoire : administrer une solution glucosée dčs que la période de jeűne débute ; en cas de chirurgie élective, arręter l'apport en protéines 24 ŕ 48 h avant l'intervention et compenser l'apport calorique avec glucose et lipides ;
- aspirer l'estomac pour éviter un apport protéique par voie digestive en cas de chirurgie oů du sang peut ętre dégluti (ORL, stomatologie) ;
- monitorage particulier : NH4 (nl : < 50 µmol/L ou 20-80 µg/dL), glycémie ;
- prévoir une anesthésie qui diminue la réaction de stress : ALR, morphiniques, analgésie postopératoire de qualité ; éviter la dexaméthasone qui augmente le catabolisme protéique ; utilisation raisonnée des antinauséeux car ils pourraient masquer les premiers signes d’encéphalopathie.
- ajouter du benzoate de Na (0,25 g/kg/j) et du phénylbutyrate de Na (0,25 g/kg/j) qui favorisent des voies métaboliques alternatives et diminuent la synthčse de NH4, et de l'HCl d'arginine (0,1 ŕ 0,2 g/kg/j) aux solutions IV ; ces solutions peuvent provoquer une hypokaliémie
- en cas d'hyperammoniémie > 3 x la normale : glucose 10 ou 20 % en IV, et dose de charge de benzoate de Na (0,25g/kg) et phénylbutyrate de Na (0,25g/kg), et selon le déficit spécifique : HCl d'arginine (0,2 g/kg) et carnitine (0,2 mg/kg/j) en IV ;
- en cas d'échec du traitement médical : hémofiltration ou dialyse péritonéale
Références :
- Dobbelare D, Mention K.
Déficits du cycle de l’urée
in Maladies métaboliques héréditaires, éditeurs B Chabrol et P de Lonlay, Progrčs en pédiatrie n°29, p139-147, Doin 2011.
- Del Rio C, Martin-Hernandez E, Ruiz A, Quijada-Fraille P, Rubio P.
Perioperative management of children with urea cycle disorders.
Pediatr Anesth 2020; 30:780-91.
Mise-à-jour août 2021