Incidence de 5-6/100.000 naissances vivantes jusqu'ŕ 32/100.000 en Polynésie française. La pathogénie en est mal comprise et probablement non univoque : anomalie du développement des canalicules biliaires, présence d'acides biliaires anormaux, infection virale périnatale (virus respiratoire syncitial, cytomégalovirus, virus d'Epstein-Barr, réovirus, rotavirus…), des facteurs génétiques ou encore une affection auto-immune.
On distingue :
- une forme embryonnaire ou malformative (10-20 %), oů l’atrésie est associée ŕ une polysplénie et ou ŕ un situs inversus avec absence de veine cave rétro-hépatique et hypoplasie de la veine porte, parfois appelée syndrome BASM (voir ce terme)
- une forme périnatale dont la cause serait infectieuse (virus ?), métabolique ou liée ŕ une malformation limitée ŕ la plaque hilaire.
Présentation clinique : ictčre néonatal ŕ bilirubine conjuguée qui se prolonge, avec selles décolorées (« mastic ») et urines sombres. Il existe une hépatomégalie, souvent accompagnée d'une splénomégalie. L'ictčre s'aggrave tandis que s'installe progressivement une cirrhose biliaire. La survenue d'une ascite et de signes d'hypertension portale est habituellement retardée, aprčs le 6čme mois de vie.
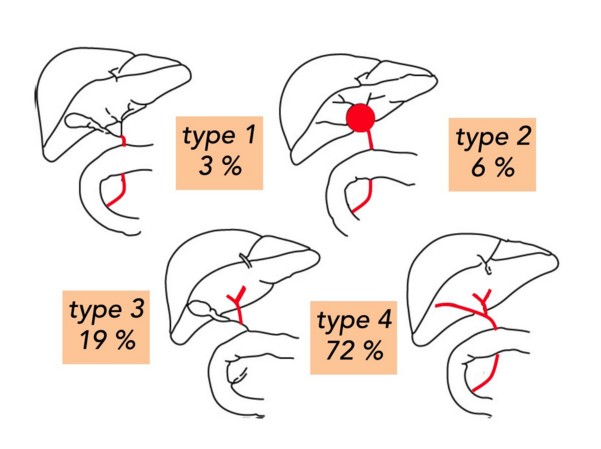
On distingue 4 types d’atrésie des voies biliaires: type 1 (3 %): atrésie du cholédoque; type 2 (6 %): kyste hilaire communicant avec des voies biliaires intra-hépatiques dystrophiques; type 3 (19 %): vésicule biliaire, cystique et cholédoque perméables; type 4 (72 %): atrésie complčte de la voie biliaire extra-hépatique.
Certaines équipes préconisent une biopsie percutanée ŕ l'aiguille ou une cholangiographie percutanée pour confirmer le diagnostic et éviter une laparotomie inutile ŕ un nouveau-né souffrant d’une hépatite néonatale, d’une maladie métabolique, d’un syndrome d’Alagille ou d’une cholestase intrahépatique progressive familiale. Le traitement habituel est l’opération de Kasaď ou porto-entérostomie pour drainer la bile dans une anse intestinale en Y et essayer de ralentir l’évolution cirrhogčne. On peut aussi réaliser : en cas de type 1: cholécysto-entérostomie, or hepatico-entérostomie ; de type 2 : kysto-entérostomie aprčs avoir vérifié par cholangiographie que le kyste hilaire communique avec les voies biliaires intrahépatiques dystrophiques ; en cas de type 3: hépatoporto-cholécystostomie.
Le pronostic de l’intervention de Kasai est meilleur si l’enfant est opéré avant l’âge de 2 mois et si l’opérateur a une large expérience de l’intervention. Plus de 50 % des enfants développe une insuffisance hépatique chronique dont le seul traitement efficace possible est la greffe hépatique. En cas de succčs, le pronostic ŕ long terme est bon : le taux de survie ŕ 10 ans est > 80 %, proche parfois de 95 %
Męme si le drainage biliaire est efficace, la fibrose déjŕ présente au moment de l’intervention de Kasaď continue ŕ évoluer lentement vers la cirrhose. Cette évolution est accélérée en cas de cholangite. Dans ce cas, l’enfant va présenter des cholangites répétées et/ou une hypertension portale progressive avec ascite et varices śsophagiennes, entraînant une détérioration progressive de la fonction hépatique et un retard de croissance. Il arrive aussi que ces enfants développent un syndrome hépato-pulmonaire ou une hypertension porto-pulmonaire(voir ces termes)
Implications anesthésiques:
- période néonatale :vérifier la fonction hépatique ; elle est en général encore normale au moment de l’intervention de Kasaď ; et l’association d’une anesthésie générale et d’une analgésie épidurale thoracique donne d’excellents résultats.
- plus tard : vérifier la fonction hépatique et le bilan de coagulation; risque d’hypertension portale; risque d’hypoplaquettose en cas de splénomégalie : malnutrition et ostéopénie fréquentes si des suppléments de vitamines ADEK et nutritionnels ne sont pas administrés
- échographie cardiaque
Références :
- Chardot C, Carton M, Spire-Bendelac N, Le Pommelet C, Golmard JL, Auvert B.
Epidemiology of biliary atresia in France: a national study 1986-96.
J Hepatol 1999; 31:1006-13.
- Kelly DA, Davenport M.
Current management of biliary atresia.
Arch Dis Child 2007; 92:1132-5.
- Khalil BA, Perera MT, Mirza DF.
Clinical practice: management of biliary atresia.
Eur J Pediatr 2010; 169:395-402.
- Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G et al.
The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study.
J Pediatr 2006; 149:393-400.
Mise à jour: février 2021